一类聚集诱导发光和压致变色发光的近红外离子型铱(Ⅲ)配合物及应用的制作方法
本发明涉及一类具有聚集诱导磷光发射和压致变色发光双重特性的离子型铱(iii)配合物近红外发光材料,及其在有机发光二极管(oleds)和应力传感器中的应用,属于发光材料领域。
背景技术:
近年来,近红外(700-2500nm)发光材料因其在信息安全显示、夜视设备、生物成像、光纤通信和医疗诊断等领域的潜在应用而备受关注。目前,已报导的近红外发光材料大致包括无机发光材料、有机发光材料、有机无机复合发光材料三大种类。其中,近红外有机发光材料主要分为有机小分子荧光材料和有机金属配合物磷光材料。与传统的小分子荧光材料相比,有机金属配合物由于能同时捕获25%的单线态激子和75%的三线态激子,理论内量子效率达100%,被认为是一类很有发展前景的近红外发光材料。目前,有机金属配合物近红外发光材料,主要包括镧系金属配合物和过渡金属配合物。其中,镧系金属配合物,如er,nd和yb,发光呈线状,发光寿命达到毫秒级,但是其特有的f-f跃迁禁阻发光特性,妨碍了镧系金属配合物量子效率的提升。相对而言,过渡金属配合物,由于重原子效应导致的强烈自旋轨道耦合作用,具有相对较高的量子效率,通过对金属原子和配体结构的改变,这类配合物可以呈现不同的发光颜色和发光效率。目前有机金属配合物近红外发光材料的研究主要集中在铂、铱、锇、铼、铷等金属配合物,其中,铂配合物近红外发光材料的电致发光效率最高。然而,铂(ii)配合物为平面四方形构型,激发态寿命长,分子易聚集,易发生三线态激子湮灭和器件效率滚降现象。与铂(ii)配合物不同,金属铱(iii)配合物为六配位的八面体构型,分子聚集的机会大大降低,能较好地抑制激子的三线态湮灭现象以及器件发光效率在高电流密度下的滚降现象(roll-off)。[1-7]
聚集荧光猝灭(aggregation-causedquenching,acq)效应是导致有机发光材料发光效率降低、并制约有机发光材料应用的主要因素。为获得有机发光材料固态下的高效发光,2001年,唐本忠课题组[8]首次报道了聚集诱导发光(aggregation-inducedemission,aie)有机荧光材料,极大拓宽了有机发光材料的发展空间,目前,基于aie特性的有机发光材料已在生化传感、生物成像、光电材料等诸多领域得到了广泛应用,并呈现了很好的商业应用前景。
典型的aie有机发光材料是四苯乙烯衍生物,但是,这些材料属于传统的荧光发光材料,发光内量子效率理论极限是25%。近年来,磷光发光材料在可见光区域已经取得了长足的发展,环金属铱(iii)配合物更是其中的佼佼者,在可见光区取得20%以上的外量子效率,但是,磷光材料因其较长的三线态激子寿命,在高电流密度下极易发生三线态-三线态湮灭(triplet-tripletannihilation,tta)和三线态极化子湮灭(triplet-polaronannihilation,tpa),导致效率滚降现象严重。为了缓解这个矛盾,磷光材料大部分都以较低的浓度掺杂于主体材料中。2007年,李富友等报导了一类具有聚集诱导磷光发射(aggregation-inducedphosphorescentemission,aipe)现象的环金属铱(iii)配合物,开启了aipe材料的研究。[9]越来越多具有aipe现象的环金属铱(iii)配合物被开发出来并应用到化学传感器、生物成像、有机电致发光器件等领域。在电致发光领域,具有aipe现象的环金属铱(iii)配合物跟常规的铱(iii)配合物相比具有显著的优点,它即使在非掺杂器件中也可以有效抑制发光器件的效率滚降现象,简化了器件的制备工艺,同时可以大幅度降低器件的制作成本,为oleds的广泛应用开辟了新的途径。
压致变色发光材料(piezochromicluminescentmaterials)是因受到外力刺激(如压力、剪切力、拉伸力等)作用下导致发光行为改变的一种新型功能材料。这种材料在信息记录与存储、力学传感器、记忆芯片和发光器件等领域拥有巨大的应用价值。近来,一些兼具聚集诱导发光(aie)和压致变色发光性质的压致变色聚集诱导发光(paie)材料,引起了研究者的极大兴趣。与传统的压致变色发光材料不同,paie材料的响应灵敏度更高,适用范围更宽广,在有机发光二极管、有机场效晶体管、传感器件等领域的应用将更为广泛。[10]
参考文献
[1]wang,zy.crcpress/taylor&francisgroup,2013.
[2]qian,g;wang,zy.chemistryanasianjournal,2010,5,1006.
[3]xiang,h;cheng,j;ma,x;etal.chemicalsocietyreviews,2013,42,6128.
[6]liao,jl;chi,y;yeh,cc;etal.journalofmaterialschemistryc,2015,3,4910.
[7]cao,x;miao,j;zhu,m;etal.chemistryofmaterials,2015,27,96.
[8]luo,jd;tang,bz;etal.chemicalcommunications,2001,0,1740.
[9]zhang,q;li,fy;etal.chemicalcommunications,2008,0,685.
[10]shan,gg;su,zm;etal.journalofmaterialschemistryc,2013,1,1440.
技术实现要素:
针对目前环金属铱(iii)配合物近红外发光材料种类少、发光效率普遍较低、且需低浓度掺杂于主体材料之中等问题,本发明致力于开发一类基于吡嗪并[2,3-f][1,10]邻菲啰啉衍生物作为受电子单元(a)、富电子的芳胺作为供电子单元的d-a(donor-acceptor)型n^n辅助配体及其离子型铱(iii)配合物,该类配合物具有聚集诱导磷光发射和压致变色发光双重特性。此类铱(iii)配合物的分子结构特点如下:(1)d-a型n^n辅助配体是以刚性稠杂环化合物吡嗪并[2,3-f][1,10]邻菲啰啉(dppz)衍生物为作为受电子单元,以芳胺类化合物作为推电子(d)单元,此类d-a构型配体引入到环金属铱(iii)配合物之中可以大幅度降低目标配合物的带隙,获得近红外发射。(2)采用刚性稠杂环dppz衍生物作为环金属铱(iii)配合物的发光核心,芳胺类化合物作为外围立体位阻单元,在保证较大的辐射跃迁速率的同时又可有效较低分子间的相互作用,缓解浓度淬灭效应,提高配合物的发光效率。(3)引入具有aie效应的芳胺结构,可以获得具有aipe特性的近红外发光材料和刺激相应的智能发光材料。
本发明开发的离子型铱(iii)配合物近红外发光材料合成条件温和、产率高、良好的成膜性和溶解性,可通过简单的溶液加工制作高效聚合物近红外发光二极管。
此类离子型铱(iii)配合物近红外发光材料的分子结构具有式1的结构:
其中r1,r2可以分别独立的选自氢原子、氰基、氟原子、三氟甲基、碳原子数为1~16的烷基、碳原子数为1~16的烷氧基;x-可以分别独立的选自f-、cl-、br-、i-、pf6-、bf4-;
式1中ar为四元取代芳环或芳杂环,d为三苯胺、咔唑、吖啶中的任何一种,ar及d单元的结构特征如式2所示:
r为碳原子数为1~16的烷基、碳原子数为1~16的烷氧基。
优选的方案是:此类离子型铱(iii)配合物近红外发光材料可以是下列式3的任何一种:
其中,r为c1~c16的带支链或直链烷烃。
为了获得上述离子型铱(iii)配合物近红外发光材料,本发明优选的合成方案如下:通过suzuki-coupling反应、schiff-base缩合反应获得相应的d-a构型的n^n配体,最终通过桥连及去桥连反应获得离子型铱(iii)配合物近红外发光材料。
本发明还提供此类阳离子型铱(iii)配合物近红外发光材料在聚合物近红外电致发光器件中的应用,将其作为单一活性发光材料应用于制备近红外电致发光器件发光层,及其高效近红外聚合物发光器件。
聚合物近红外电致发光器件的结构包括氧化铟锡(ito)导电玻璃衬底阳极,空穴注入层,发光层,电子传输层和阴极;其中,空穴注入层为聚二氧乙基噻吩以及聚苯乙烯磺酸(pedot:pss)涂层,空穴传输层为聚[双(4-苯基)(4-丁基苯基)胺](poly-tpd)涂层,发光层为单一发光材料和主体材料的共混涂层或者不掺主体的单发光层,1,3,5-三(1-苯基-1h-苯并咪唑-2-基)苯(tpbi)为电子传输层,氟化铯(csf)为电子注入层,阴极为铝的沉积层。
主体材料由聚(9-乙烯咔唑)(pvk)和2,2'-(1,3-苯基)二[5-(4-叔丁基苯基)-1,3,4-噁二唑](oxd-7)组成,它们的质量分数为pvk:oxd-7=7:3;发光层中发光材料与主体材料的质量百分比分别为70~100%,0~30%。
附图说明
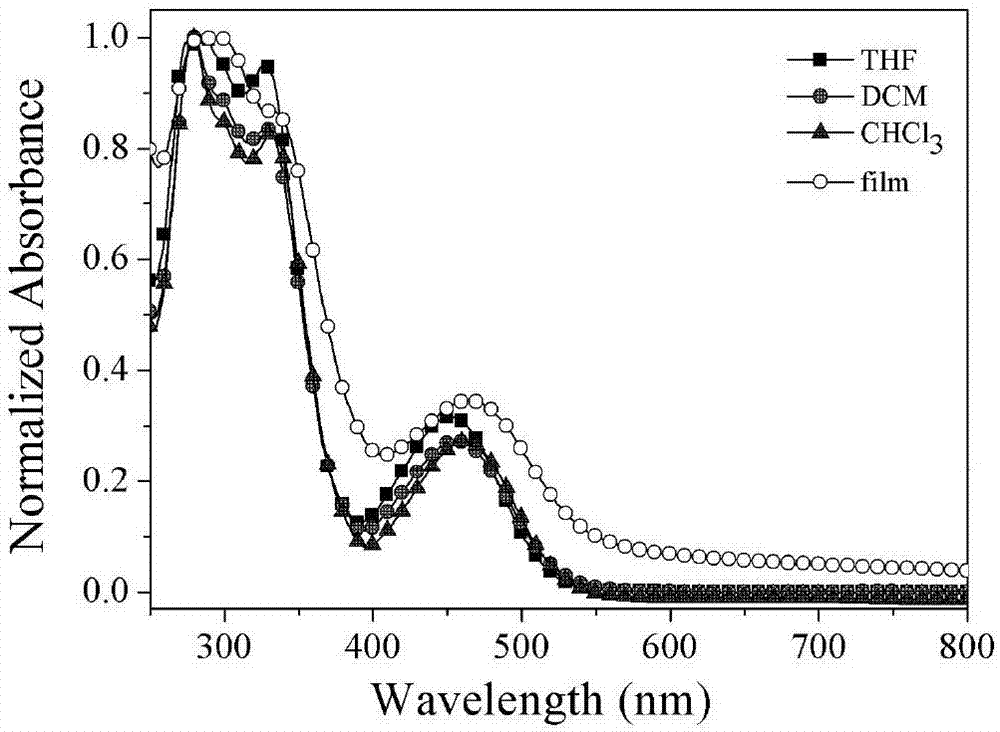
图1为本发明实施例1中n^n配体2的紫外-可见吸收光谱图。
图2为本发明实施例1中n^n配体2的光致发光光谱图。
图3为本发明实施例1中配合物(2fppy)2ir+[dppz-(11,12-ditpa)2]pf6﹣的紫外-可见吸收光谱图。
图4为本发明实施例1中配合物(2fppy)2ir+[dppz-(11,12-ditpa)2]pf6﹣的光致发光光谱图。
图5为本发明实施例2中配合物(ppy)2ir+[dppz-(11,12-ditpa)2]pf6﹣的紫外-可见吸收光谱图。
图6为本发明实施例2中配合物(ppy)2ir+[dppz-(11,12-ditpa)2]pf6﹣的光致发光光谱图。
图7为本发明实施例1中配合物(2fppy)2ir+[dppz-(11,12-ditpa)2]pf6﹣在不同水含量的thf/h2o混合溶剂中的光致发光光谱及其相对强度与水含量的曲线图。
图8为本发明实施例1中配合物(2fppy)2ir+[dppz-(11,12-ditpa)2]pf6﹣在不同水含量的thf/h2o混合溶剂中的发光强度与水含量的曲线图。
图9为本发明实施例2中配合物(ppy)2ir+[dppz-(11,12-ditpa)2]pf6﹣在不同水含量的thf/h2o混合溶剂中的光致发光光谱及其相对强度与水含量的曲线图。
图10为本发明实施例2中配合物(ppy)2ir+[dppz-(11,12-ditpa)2]pf6﹣在不同水含量的thf/h2o混合溶剂中的发光强度与水含量的曲线图。
图11为本发明实施例1中配合物(2fppy)2ir+[dppz-(11,12-ditpa)2]pf6﹣在研磨前与研磨后的光致发光光谱图。
图12为本发明实施例2中配合物(ppy)2ir+[dppz-(11,12-ditpa)2]pf6﹣在研磨前与研磨后的光致发光光谱图。
图13为本发明实施例1中配合物(2fppy)2ir+[dppz-(11,12-ditpa)2]pf6﹣在掺杂与非掺杂条件下的电致发光光谱图(el)及外量子效率-电流密度曲线(eqe-j)图。
图14为本发明实施例1中配合物(2fppy)2ir+[dppz-(11,12-ditpa)2]pf6﹣在掺杂与非掺杂条件下的外量子效率-电流密度曲线(eqe-j)图。
图15为本发明实施例2中配合物(2fppy)2ir+[dppz-(11,12-ditpa)2]pf6﹣在掺杂与非掺杂条件下的电致发光光谱图(el)及外量子效率-电流密度曲线(eqe-j)图。
图16为本发明实施例2中配合物(2fppy)2ir+[dppz-(11,12-ditpa)2]pf6﹣在掺杂与非掺杂条件下的外量子效率-电流密度曲线(eqe-j)图。
具体实施方案
以下具体实施例旨在对本发明作进一步说明,但这些具体实施方案不以任何方式限制本发明的保护范围。
实施例1
离子型铱(iii)配合物(2fppy)2ir+[dppz-(11,12-ditpa)2]pf6﹣的制备
该铱(iii)配合物的合成路线如下:
中间体1的合成
在100ml双口瓶中,依次加入4-硼酸三苯胺(1.40g,4.70mmol)、4,5-二溴邻苯二胺(0.50g,1.88mmol)、四(三苯基膦)合钯(130mg,0.029mmol)、2mol/l碳酸钾溶液(8ml)、甲苯(20ml),氮气氛围下,加热至80℃,搅拌反应12h。冷却至室温,减压蒸馏除去甲苯,剩余液用二氯甲烷萃取、水洗、无水mgso4干燥、过滤,减压蒸馏除去溶剂,真空干燥3h,得白色固体894mg,产率80%。因产物对氧气极其敏感,故未经纯化直接投入下一步反应。
中间体2(n^n配体)的合成
将中间体1(916mg,1.50mmol)、1,10-菲啰啉-5,6-二酮(316mg,1.50mmol)和无水乙醇(30ml)、醋酸(1ml)加入到100ml圆底烧瓶中,加热至80℃,搅拌反应4h,产生大量砖红色固体,冷却至室温,抽滤,滤饼经无水乙醇(10×3ml)洗涤后用中性三氧化二铝柱色谱分离,洗脱剂为二氯甲烷和乙醇的混合溶液(50/1,v/v),得砖红色固体1.04g,产率90%。1hnmr(400mhz,cdcl3)δ(ppm):9.63(d,j=7.9hz,2h),9.27(d,j=3.1hz,2h),8.37(s,2h),7.80(dd,j=8.1,4.4hz,2h),7.31–7.26(m,10h),7.21(s,2h),7.15(d,j=7.7hz,8h),7.06(t,j=7.5hz,8h).maldi-tof-ms(m/z):calcdforc54h36n6:768.92;found,769.98[m+1]+.
中间体3的合成
在100ml单口瓶中,加入2-(2,4-二氟苯基)吡啶(1.0g,5.23mmol)、水合三氯化铱(ircl3·nh2o)(777.8mg,2.61mmol)、蒸馏水(5ml)和乙二醇单乙醚(15ml),氮气保护下加热100℃,搅拌反应18h。冷却反应液至室温,加入蒸馏水(5ml),析出大量黄绿色固体,抽滤,固体依次用蒸馏水、石油醚和正己烷洗涤,真空干燥,得黄绿色粉末产物1.30g,产率82%。
配合物(2fppy)2ir+[dppz-(11,12-ditpa)2]pf6﹣的合成
在100ml单口瓶中,依次加入中间体3(100mg,0.08mmol)、中间体2(123mg,0.16mmol)及二氯甲烷与甲醇的混合溶剂24ml(3/1,v/v),氮气保护下,加热40℃,搅拌反应12h,冷却至室温,加入六氟磷酸钾(30mg,0.16mmol),室温下继续搅拌反应4h,反应结束后抽滤,滤饼用二氯甲烷洗涤(10×2ml),收集有机滤液,干燥,蒸馏出溶剂,剩余物通过中性三氧化二铝柱色谱分离,洗脱剂为二氯甲烷和乙醇的混合溶液(10/1,v/v),得橙红色产物180mg,产率76%。1hnmr(400mhz,cdcl3)δ(ppm):9.88(dd,j=8.2,1.4hz,2h),8.42(s,2h),8.33(dt,j=8.0,4.1hz,4h),8.00(dd,j=8.3,5.2hz,2h),7.77(t,j=8.2hz,2h),7.59(d,j=5.7hz,2h),7.32–7.26(m,8h),7.21(d,j=8.6hz,4h),7.15(d,j=7.6hz,8h),7.09–7.03(m,10h),6.67–6.60(m,2h),5.80(dd,j=8.2,2.3hz,2h).maldi-tof-ms(m/z):calcdfor[m-pf6]+c76h48f4irn8:1341.49;found,1341.75.
实施例2
离子型铱(iii)配合物(ppy)2ir+[dppz-(11,12-ditpa)2]pf6﹣的制备
该配合物的合成路线如下:
中间体4的合成
在100ml单口瓶中,依次加入2-苯基吡啶(1.0g,6.44mmol)、水合三氯化铱(ircl3·nh2o)(959.6mg,3.22mmol)、蒸馏水(5ml)和乙二醇单乙醚(15ml),在氮气保护下,加热至100℃,搅拌反应24h。冷却反应体系至室温,加入蒸馏水(5ml),析出大量黄固体,抽滤,固体依次用蒸馏水、石油醚和正己烷洗涤多次,真空干燥得黄色粉末产物1.50g,产率87.0%。
配合物(ppy)2ir+[dppz-(11,12-ditpa)2]pf6﹣的合成
在100ml单口瓶中,依次加入中间体4(100mg,0.09mmol)、中间体2(138mg,0.18mmol)及二氯甲烷与甲醇的混合溶剂xxml(3/1,v/v),氮气保护下,加热至40℃,搅拌反应12h。冷却至室温,加入六氟磷酸钾(33mg,0.16mmol),室温下继续反应4h,反应结束后抽滤,滤饼用二氯甲烷洗涤(10×2ml),收集有机滤液,干燥,蒸馏出溶剂,剩余物通过中性三氧化二铝柱色谱分离,洗脱剂为二氯甲烷和乙醇的混合溶液(20/1,v/v),得暗红色产物190mg,产率75%。1hnmr(400mhz,cdcl3)δ(ppm):9.85(dd,j=8.2,1.4hz,2h),8.45(s,2h),8.37(dd,j=5.1,1.4hz,2h),7.97(dd,j=8.2,5.1hz,4h),7.76(t,j=7.0hz,4h),7.63(d,j=5.5hz,2h),7.36–7.30(m,8h),7.25(d,j=8.6hz,4h),7.23–7.05(m,18h),7.03(t,j=6.8hz,4h),6.46(d,j=7.3hz,2h).maldi-tof-ms(m/z):calcdfor[m-pf6]+c76h52irn8:1269.52;found,1270.09.
实施例3
11,12-二(4-三苯胺基)二吡啶并[3,2-a:2',3'-c]吩嗪的光物理性能
中间体2为11,12-二(4-三苯胺基)二吡啶并[3,2-a:2',3'-c]吩嗪n^n配体,其紫外-可见(uv-vis)吸收光谱及荧光光谱(pl)分别如附图1和图2所示。将此n^n配体配制成1×10-5mol/l的四氢呋喃(thf)、三氯甲烷(chcl3)、二氯甲烷(dcm)溶液,并依次移取3ml置于石英比色皿中,测试这些溶液的uv-vis和pl光谱;配制10mg/ml的chcl3溶液,通过旋转涂敷于石英片上,测试这些薄膜的uv-vis和pl光谱。
由图1可以看出:(1)配体在250nm至550nm均有吸收并呈现两个特征吸收带,其中,250nm到400nm的强吸收带归属于π→π*的电荷转移跃迁吸收,而450nm至550nm的中强吸收带归属于配体内的电荷转移跃迁吸收(ict);(2)随着溶剂极性的增大,ict吸收带有略微的红移,说明该配体的基态受溶剂极性的影响较小;(3)在固体薄膜中,ict吸收带呈现明显的红移,说明在固体薄膜中,n^n配体存在较强的分子间相互作用。
由图2可以看出:n^n配体在溶液状态和固体状态下,都有强烈的荧光发射。由于配体具有明显的ict性质,因此,随着溶剂极性的增大,配体的发射光谱逐渐红移并变成无精细结构的宽峰。配体在二氯甲烷、三氯甲烷、四氢呋喃中的发射峰依次是654、631、625nm;但是在固体薄膜中,因为溶剂化显色效应的消除,配体的荧光发射峰相对极性溶剂如二氯甲烷有明显的蓝移,蓝移至614nm。
实施例4
离子型铱(iii)配合物(2fppy)2ir+[dppz-(11,12-ditpa)2]pf6﹣的光物理性能
离子型铱配合物(2fppy)2ir+[dppz-(11,12-ditpa)2]pf6﹣在室温条件下的吸收光谱及发射光谱分别如附图3和4所示。将配合物(2fppy)2ir+[dppz-(11,12-ditpa)2]pf6﹣配制成1×10-5mol/l的四氢呋喃(thf)、三氯甲烷(chcl3)、二氯甲烷(dcm)溶液,并移取3ml置于石英比色皿中,测试这些离子型铱配合物的uv-vis和pl光谱;配制10mg/ml的chcl3溶液,通过旋转涂敷于石英玻璃片上,并测试其uv-vis和pl光谱,配合物的发光行为因受氧气影响,pl测试需在无氧条件下进行。
由图3可见:这类离子型铱配合物的吸收光谱亦呈现明显的两个特征峰,并相对于自由的n^n配体有明显的红移。其中,小于400nm的高能吸收带归属于配体内部自旋允许的π→π*的电荷转移跃迁吸收,425nm至650nm范围的中强吸收带归属于配体的ict吸收以及金属到配体的电荷转移跃迁(1mlct)混合态吸收。
由图4可见:配合物在溶液中的发光表现出明显的溶剂化显色现象。在极性小的溶剂中如三氯甲烷和四氢呋喃,配合物呈现较窄的发射峰,最大发射峰分别是700、743nm,而在极性较大的溶剂中时表现较大的发光红移,如在二氯甲烷中,配合物的发射峰值是775nm。在固体薄膜状态时,配合物的最大发射峰与极性溶剂中相比呈现较大的蓝移,这来源于溶剂化显色效应的消除。
实施例5
离子型铱(iii)配合物(ppy)2ir+[dppz-(11,12-ditpa)2]pf6﹣的光物理性能
将离子型铱配合物(ppy)2ir+[dppz-(11,12-ditpa)2]pf6﹣分别配制成1×10-5mol/l的四氢呋喃(thf)、三氯甲烷(chcl3)、二氯甲烷(dcm)溶液,并移取3ml置于石英比色皿中,测试其uv-vis和pl光谱,配制10mg/ml的chcl3溶液,通过旋转涂敷于石英玻璃片上,测试其固体薄膜的uv-vis和pl光谱如附图5和6所示。
由图5可见:离子型铱配合物(ppy)2ir+[dppz-(11,12-ditpa)2]pf6﹣的吸收光谱亦呈现明显的两个特征峰,并相对于自由的n^n配体有明显的红移。其中,小于450nm的高能吸收带归属于配体内部自旋允许的π→π*的电荷转移跃迁吸收,450nm至650nm范围的中强吸收带归属于配体的ict吸收以及金属到配体的电荷转移跃迁(1mlct)混合态吸收。
由图6可见:离子型铱配合物(ppy)2ir+[dppz-(11,12-ditpa)2]pf6﹣在四氢呋喃、三氯甲烷、二氯甲烷中的发射峰依次是750、700、770nm,呈现明显的溶剂化显色现象,而在纯固体薄膜中的最大发射峰为743nm,相对溶液中的发射光谱蓝移,归因于固体薄膜中溶剂化效应的消除。
实施例6
离子型铱(iii)配合物(2fppy)2ir+[dppz-(11,12-ditpa)2]pf6﹣的aipe性能
配制1×10-4mol/l的四氢呋喃溶液,取原始溶液1ml于10ml容量瓶中,加入体积比为10~90%的水,最终补加thf至浓度1×10-5mol/l,摇匀后超声0.5h,测试配合物在不同体积水含量的thf/h2o混合溶剂中的光致发光行为,其光致发光光谱如附图7所示,发光强度相对水含量的曲线图如附图8所示。从图7、8可知,在水含量低于80%时,配合物发光比较微弱;当水体积进一步增大,超过80%以上时,配合物呈现明显的磷光发射增强现象;当水含量为90%时,配合物/thf/h2o混合液的发光强度比配合物/thf溶液的发光强度增强了25倍,表现出明显的aipe性质,且最大发射峰位于709nm,为近红外发射。我们认为,在溶液状态下,分子的自由度较大,由于双取代三苯胺的作用,激发态的能量大都以振动弛豫方式损失,在聚集状态下,分子间的距离减小,分子的自由度受到限制,分子的扭转、振动减小,激发态结构弛豫变小,辐射跃迁速率增大,磷光发射强度大幅度增强,表现出aipe的性能。
实施例7
离子型铱(iii)配合物(ppy)2ir+[dppz-(11,12-ditpa)2]pf6﹣的aipe性能
配合物(ppy)2ir+[dppz-(11,12-ditpa)2]pf6﹣的thf/h2o混合液的光致发光行为测试方法同实施例6,其光致发光光谱如附图9所示,发光强度相对水含量的曲线图如附图10所示。由图9、10可见:该配合物同样表现出明显的aipe的现象。当水含量为90%时,配合物/thf/h2o混合液的发光强度比配合物/thf溶液的发光强度增强了15倍,最大发射峰位于710nm,为近红外发射。
实施例8
离子型铱(iii)配合物(2fppy)2ir+[dppz-(11,12-ditpa)2]pf6﹣的压致变色发光性能
附图11为配合物(2fppy)2ir+[dppz-(11,12-ditpa)2]pf6﹣在研磨前和研磨后的光致发光光谱图。结果表明,未研磨时,配合物发射强烈的饱和红光,发射峰值620nm;研磨后,外观由砖红色转变成暗红色,发光光谱发生了106nm的红移,最大发射峰红移至726nm,为一类性能突出的近红外压致变色材料。我们认为,11,12-二(4-三苯胺基)二吡啶并[3,2-a:2',3'-c]吩嗪n^n配体[dppz-(11,12-ditpa)2]的大刚性平面结构是压致变色行为的重要来源。在研磨之前,配合物为松散的晶型结构,受到外力挤压后,由晶型结构向不定型结构转变,且平面之间的π-π堆积作用较晶型状态时更强,因此,其光致发光行为表现为大幅度的红移。
实施例9
离子型铱(iii)配合物(ppy)2ir+[dppz-(11,12-ditpa)2]pf6﹣的压致变色发光性能
附图12为配合物(ppy)2ir+[dppz-(11,12-ditpa)2]pf6﹣在研磨前和研磨后的光致发光光谱图。该配合物亦表现出明显的压致变色性能。研磨前,配合物发射强烈的深红光,发射峰值为650nm,研磨之后,配合物的发射峰值为760nm,发生110nm的红移。如此大幅度的红移现象,在应力传感器等领域具有极大的商业应用价值。
实施例10
实施例1中离子型铱(iii)配合物(2fppy)2ir+[dppz-(11,12-ditpa)2]pf6﹣的聚合物近红外电致发光器件性能
聚合物电致发光器件的掺杂器件结构为ito(110nm)/pedot:pss(40nm)/poly-tpd(30nm)/pvk:oxd(7:3):dopant,50nm)/tpbi(60nm)/csf(0.8nm)/al(120nm);非掺杂器件结构为ito(110nm)/pedot:pss(40nm)/pvk(30nm)/dopant(40nm)/tpbi(60nm)/csf(0.8nm)/al(120nm)。其中,氧化铟锡(ito)玻璃基底为阳极,聚二氧乙基噻吩(pedot)和聚苯乙烯磺酸钠(pss)为空穴注入层,聚[双(4-苯基)(4-丁基苯基)胺](poly-tpd)和聚乙烯基咔唑(pvk)分别为空穴传输层,1,3,5-三(1-苯基-1h-苯并咪唑-2-基)苯(tpbi)为电子传输层,氟化铯(csf)为电子注入层,阴极为铝的沉积层。在掺杂器件中,主体材料是由pvk和2,2'-(1,3-苯基)二[5-(4-叔丁基苯基)-1,3,4-噁二唑](oxd-7)组成(质量比为7:3),发光层是主体材料和配合物客体材料(dopant)(2fppy)2ir+[dppz-(11,12-ditpa)2]pf6﹣的共混涂层,掺杂配合物的质量百分数为70%。在非掺杂器件中,发光层为单一的配合物(2fppy)2ir+[dppz-(11,12-ditpa)2]pf6﹣涂层。
聚合物电致发光器件的电致发光光谱图(el)、外量子效率-电流密度曲线(eqe-j)分别如附图13和14所示。由图可见:在掺杂器件中,器件的最大电致发光峰位于702nm,eqemax达到1.27%,最大辐照度(rmax)为60μw/sr/cm2。在非掺杂的器件中,器件的最大电致发光波长为738nm,eqemax为0.49%,最大辐照度(rmax)为237μw/sr/cm2。值得注意的是,以该配合物为发光层的聚合物电致发光器件都呈现出较小的效率滚降。
实施例11
实施例2中离子型铱(iii)配合物(ppy)2ir+[dppz-(11,12-ditpa)2]pf6﹣的聚合物近红外电致发光器件性能
聚合物电致发光器件的器件结构与实施例10相同。聚合物电致发光器件的电致发光光谱图(el)、外量子效率-电流密度曲线(eqe-j)分别如附图15和16所示。由图可见:在掺杂器件中,器件的最大电致发光峰位于704nm,eqemax达到0.92%,最大辐照度(rmax)为53μw/sr/cm2。在非掺杂器件中,器件的最大电致发光波长为746nm,eqemax为0.19%,最大辐照度(rmax)为47μw/sr/cm2。值得注意的是,以该配合物为发光层的聚合物电致发光器件也都呈现出较小的效率滚降。
尽管结合了优选实施例对本发明进行了说明,但本发明并不局限于上述实施案例,应当理解所附权利要求概括了本发明的范围。在本发明构思的指导下,本领域的技术人员应当意识到,对本发明的各实施例方案所进行的一定的改变,都将被本发明的权利要求书的精神和范围所覆盖。
- 还没有人留言评论。精彩留言会获得点赞!