一种4-(2-噻吩基)三苯胺及其衍生物、制备方法与应用与流程
本发明涉及紫外线吸收剂及其制备领域,更具体地,涉及一种4-(2-噻吩基)三苯胺及其衍生物、制备方法与应用。
背景技术:
紫外线吸收剂是能够吸收由太阳辐射出来和人造光源中所产生的紫外线,而自身不受影响的一种光稳定剂。在紫外线的作用下,由于高分子材料吸收了紫外线的能量,高分子材料会发生自动氧化反应,从而导致了聚合物发生降解,使得其外观和使用性能下降;而如果在高分子材料的生产或者加工成型过程中添加紫外线吸收剂,其能吸收紫外线,转化成对高分子材料无害的能量而释放。由于高分子材料聚合物的种类不同,引起其老化的紫外线的波长也不同,因此在添加紫外线吸收剂时,应根据不同的聚合物类型,选择相应的紫外线吸收剂。
紫外线吸收剂应该具有如下特点:具有强烈地吸收紫外线能力,特别是对波长为290~400nm之间的紫外线;并且,能与高分子材料及其他助剂相容,不发生喷霜和析出现象;有优良的热稳定性,在材料加工过程中,不发生热分解;紫外线吸收剂自身光稳定性、化学稳定性好,不发生自分解,不变色;吸收剂自身无毒或者毒性较低,价格低廉。
专利cn201410205811.5公开了一种硫脲功能化的三苯胺类有机染料及其应用,该三苯胺类有机染料的紫外吸收范围宽,用于制备太阳能电池。目前,含有三苯胺的化合物多数用于制备太阳能电池材料或有机光电材料,还没有用于作为紫外线吸收剂的报道。因此,可以利用三苯胺较大的空间位阻、超共轭电子效应和较高的活性特点,制备一种紫外线吸收剂。
伴随着高分子材料的应用领域不断扩大,加上近些年环境被严重地破坏,紫外线的危害越来越严重,相应的紫外线吸收剂在未来必将得到更进一步的发展。目前常使用的紫外线吸收剂的紫外吸收性能较差,效率不高。
因此,需要制备一种对紫外线吸收能力较强的紫外线吸收剂。
技术实现要素:
本发明为克服上述现有技术所述紫外线吸收剂的紫外吸收性能较差的缺陷,提供一种4-(2-噻吩基)三苯胺及其衍生物。本发明提供的4-(2-噻吩基)三苯胺及其衍生物对紫外线的吸收能力较强,可以作为一种紫外线吸收剂。
本发明的另一目的在于提供上述4-(2-噻吩基)三苯胺及其衍生物的制备方法。
本发明的还一目的在于提供上述4-(2-噻吩基)三苯胺及其衍生物在紫外线吸收剂中的应用。
为解决上述技术问题,本发明采用的技术方案是:
一种4-(2-噻吩基)三苯胺及其衍生物,所述4-(2-噻吩基)三苯胺及其衍生物具有如式(i)或式(ii)所示结构:
本发明提供的4-(2-噻吩基)三苯胺及其衍生物含有噻吩基团和/或卤原子。噻吩基团可以加强4-(2-噻吩基)三苯胺及其衍生物的共轭效应,利用噻吩基团上的富电子结构,使得外界紫外线的能量被电子的迁移跃迁所消耗,一个噻吩基团可以控制4-(2-噻吩基)三苯胺及其衍生物的紫外线吸收在紫外区。并且,4-(2-噻吩基)三苯胺及其衍生物是在吸收紫外线能量后,利用自身的非辐射能量传递来消耗能量。因此,4-(2-噻吩基)三苯胺及其衍生物在紫外区具有较强的吸收能力,可以作为一种紫外线吸收剂。
优选地,所述x为溴原子或碘原子。
卤原子(溴或碘原子)的重原子效应能产生荧光猝灭,降低了式(ii)的4-(2-噻吩基)三苯胺及其衍生物的荧光强度,使其不发荧光;并且式(ii)的4-(2-噻吩基)三苯胺及其衍生物具有更为优异的紫外吸收能力。
不发荧光的优点在于:式(ii)的4-(2-噻吩基)三苯胺及其衍生物只会吸收光线中的紫外线,不会因为紫外线的激发而使原来光线发出蓝光,即式(ii)的4-(2-噻吩基)三苯胺及其衍生物能够让原来光线保持自然的光色,对透过光线没有影响。若紫外线吸收剂具有荧光,则会有蓝光发出,增加了原来光线中的蓝色成分,使光的颜色偏蓝,不仅失去了原来光线所具有的本色,而且增加了对人眼睛的伤害。
更优选地,所述x为溴原子。
本发明同时保护上述4-(2-噻吩基)三苯胺及其衍生物的制备方法,所述制备方法包括如下步骤:
s1.将三苯胺与卤化试剂进行卤化反应,得到4-卤三苯胺;
s2.将4-卤三苯胺与2-噻吩硼酸进行suzuki偶联反应,得到式(i)的4-(2-噻吩基)三苯胺及其衍生物;
s3.将4-(2-噻吩基)三苯胺与卤化试剂进行卤化反应,得到式(ii)的4-(2-噻吩基)三苯胺及其衍生物。
本发明提供的4-(2-噻吩基)三苯胺及其衍生物的制备方法是将三苯胺进行卤化反应和suzuki偶联反应。星型结构的三苯胺有着较大的空间位阻和超共轭电子效应,连着的三个苯环有着较高活性和易于被修饰的特点。由于三苯胺自由基特有的性质,使得4-(2-噻吩基)三苯胺及其衍生物有着超共轭电子效应和高的空穴迁移率。噻吩基团具有芳杂环结构,形成π-π共轭,化学稳定性好;噻吩类衍生物的吸收光谱与太阳辐射的光谱较吻合,能够较强地吸收太阳光(尤其是紫外线);当噻吩基团作为修饰基团时,可以加强修饰后化合物的共轭效应,利用噻吩基团上的富电子结构,外界紫外线的能量会被电子的迁移跃迁所消耗掉,从而可以增长化合物的紫外线吸收光谱。
优选地,所述卤化试剂为n-溴代丁二酰亚胺或n-碘代丁二酰亚胺。
优选地,步骤s1.中所述三苯胺与卤化试剂的摩尔比为1∶1.03~1.10。该范围内的摩尔比能确保反应底物三苯胺反应完全而不会出现较多的副产物二卤代产物。
更优选地,步骤s1.中所述三苯胺与卤化试剂的摩尔比为1∶1.04。
优选地,步骤s1.中所述卤化反应的反应条件为搅拌下-5~0℃冷浴8~9h。该反应条件下能确保反应完全进行。
更优选地,步骤s1.中所述卤化反应的反应条件为搅拌下-3~0℃冷浴8~9h。
进一步优选地,步骤s1.中所述卤化反应的反应条件为搅拌下0℃冷浴8h。
具体过程如下:先在反应容器单口烧瓶上覆盖一层锡纸,进行遮光处理,然后加入三苯胺,再加入三氯甲烷来溶解反应物,开启搅拌器,并保持冷浴的状态;15分钟后,使反应物以及溶剂等整个反应体系的温度降至0℃或以下,然后少量多次地在20min内加入卤化试剂,使卤化试剂的浓度保持在很低的水平,尽量减少副产物二卤代产物的产生,添加完毕后,持续搅拌并且冷浴8h。
优选地,步骤s1.中所述反应后还包括分离提纯的步骤。
更优选地,所述分离提纯的过程为:反应结束后,用旋转蒸发仪除去粗产品中的三氯甲烷,以正己烷和二氯甲烷为淋洗剂,采用硅胶层析柱分离提纯,收集产物,再用旋转蒸发仪除去淋洗剂,得到4-卤三苯胺。
优选地,所述正己烷与二氯甲烷的体积比为6∶1。
优选地,步骤s2.中所述4-卤三苯胺与2-噻吩硼酸的摩尔比为1∶2.10~2.80。4-卤三苯胺与2-噻吩硼酸的摩尔比在1∶2.10~2.80范围内,可确保反应底物4-卤三苯胺反应完全,使产物的产率最大化,并且不会浪费太多2-噻吩硼酸。
更优选地,步骤s2.中所述4-卤三苯胺与2-噻吩硼酸的摩尔比为1∶2.58。
优选地,步骤s2.中所述suzuki偶联反应选用四(三苯基膦)钯为催化剂,四氢呋喃为溶剂。
优选地,步骤s2.中所述suzuki偶联反应的条件为在氮气保护下加热搅拌;所述加热搅拌的温度为70~80℃,时间为40~55h。该温度可以使反应物充分溶解并保持较适合的反应条件,减少高温下2-噻吩硼酸的损耗;反应时间由反应过程中提取反应液进行tlc点板监控而得,一般要40小时以上,4-卤三苯胺才会反应比较完全,但反应时间过长,产生副产物的机率就会增大。
更优选地,所述加热搅拌的温度为70~75℃,时间为40~48h。进一步优选地,所述加热搅拌的温度为75℃,时间为48h。
suzuki偶联反应的具体过程如下:将4-卤三苯胺、2-噻吩硼酸和催化剂加入反应容器两口烧瓶中,加入碱性物质,再加入thf溶液,然后对反应装置进行密封处理,抽出空气,在氮气保护下,恒温搅拌进行suzuki偶联反应。
优选地,步骤s2.中所述suzuki偶联反应选用碳酸钾溶液为碱性物质。
优选地,所述碳酸钾溶液的浓度为1.90~2.10mol/l。
更优选地,所述碳酸钾溶液的浓度为2mol/l。
优选地,步骤s2.中所述反应后还包括分离提纯的步骤。
更优选地,所述分离提纯的过程为:将反应后的溶液用二氯甲烷萃取,萃取后产物用饱和食盐水洗涤,再用旋转蒸发仪除去萃取剂;再对产物以体积比为6∶1的正己烷和二氯甲烷作为淋洗剂,进行硅胶层析柱分离提纯,收集产物,再用旋转蒸发仪除去溶剂,得到式(i)的4-(2-噻吩基)三苯胺及其衍生物。
优选地,步骤s3.中所述4-(2-噻吩基)三苯胺与卤化试剂的摩尔比为1∶1.02~1.07。4-(2-噻吩基)三苯胺与卤化试剂的摩尔比在1∶1.02~1.07范围内,能确保绝大部分卤化试剂作用在噻吩基上,而不是苯环上。
更优选地,步骤s3.中所述4-(2-噻吩基)三苯胺与卤化试剂的摩尔比为1∶1.04。
优选地,步骤s3.中所述卤化反应的反应条件为搅拌下-5~0℃冷浴6~7h。该反应条件能确保卤化反应进行完全。
更优选地,步骤s3.中所述卤化反应的反应条件为搅拌下-3~0℃冷浴6~7h。
进一步优选地,步骤s3.中所述卤化反应的反应条件为搅拌下0℃冷浴6h。
具体过程如下:先在反应容器单口烧瓶上覆盖一层锡纸,进行遮光处理,然后加入三苯胺,再加入三氯甲烷来溶解反应物,开启搅拌器,并保持冷浴的状态;15分钟后,使反应物以及溶剂等整个反应体系的温度降至0℃或以下,然后少量多次地在30min内加入卤化试剂,使卤化试剂的浓度保持在非常低的水平,尽量避免在苯环上发生卤代反应,添加完毕后,持续搅拌并且冷浴6h。
优选地,步骤s3.中所述反应后还包括分离提纯的步骤。
更优选地,所述分离提纯的过程为:反应结束后,用旋转蒸发仪除去粗产品中的三氯甲烷,以正己烷为淋洗剂,采用硅胶层析柱分离提纯,收集产物,再用旋转蒸发仪除去淋洗剂,得到式(ii)的4-(2-噻吩基)三苯胺及其衍生物。
本发明还保护上述4-(2-噻吩基)三苯胺及其衍生物在制备紫外线吸收剂中的应用。
本发明提供的4-(2-噻吩基)三苯胺及其衍生物在紫外区具有较强的吸收能力,可以作为一种紫外线吸收剂。
优选地,所述紫外线吸收剂在塑料、塑料薄膜、军用机械、建筑材料、橡胶制品或化妆品中的应用。
与现有技术相比,本发明的有益效果是:
本发明提供的4-(2-噻吩基)三苯胺及其衍生物对紫外线具有较强的吸收能力,可以作为一种紫外线吸收剂;进一步地,当4-(2-噻吩基)三苯胺及其衍生物具有式(ii)所示结构的时候,卤原子的重原子效应能产生荧光猝灭,使得4-(2-噻吩基)三苯胺及其衍生物不发荧光,不会对透过光线有影响,能够使原来的光线保持本色,同时也减小了对人眼睛的伤害。
附图说明
图1为实施例1中c3的核磁共振氢谱图;
图2为实施例1中c4的核磁共振氢谱图;
图3为实施例1中c3的质谱图;
图4为实施例1中c4的质谱图;
图5为实施例1中c3的红外光谱图;
图6为实施例1中c4的红外光谱图;
图7为实施例1中c3和c4的二氯甲烷溶液的紫外可见光吸收光谱图;
图8为实施例1中c3和c4的薄膜的紫外可见光吸收光谱图;
图9为实施例1中c3和c4的二氯甲烷溶液的荧光光谱图;
图10为实施例1中c3和c4的薄膜的荧光光谱图。
具体实施方式
下面结合具体实施方式对本发明作进一步的描述,但本发明的实施方式不限于此。实施例中的原料均可通过市售得到;除非特别说明,本发明采用的试剂、方法和设备为本技术领域常规试剂、方法和设备。
实施例1
一种4-(2-噻吩基)三苯胺及其衍生物,其制备方法具体包括如下步骤:
s1.先在反应容器单口烧瓶上覆盖一层锡纸,进行遮光处理,再将0.0025mol三苯胺c1置于经过遮光处理过的单口烧瓶中,加入40ml三氯甲烷,溶解反应物,开启搅拌器,并保持0℃冷浴的状态;15分钟后,少量多次地在20min内加入n-溴代丁二酰亚胺(一般情况下,nbs可以为0.00258~0.00275mol,在实施例1中为0.0026mol),添加完毕后,持续搅拌并且冷浴(一般情况下,冷浴时间可以为8~9h,在实施例1中为8h);
8h后,利用旋转蒸发仪除去粗产品中的氯仿,以正己烷与二氯甲烷体积比为6∶1的溶液作为淋洗剂,通过硅胶层析柱分离提纯,收集产物,再用旋转蒸发仪除去淋洗剂,得到0.6742g淡黄色晶体产物c2。c2的名称为4-溴三苯胺,分子式为c18h14brn,产率为84.66%。
s2.将0.0019molc2和2-噻吩硼酸(一般情况下,2-噻吩硼酸可以为0.0040~0.0053mol,在实施例1中为0.0049mol)加入反应容器两口烧瓶中,加入0.000098mol催化剂四(三苯基膦)钯,加入2mol/l的k2co3溶液10ml,最后加入50mlthf溶液,对反应装置进行密封处理,抽出空气,在氮气保护下,恒温75℃并持续搅拌,进行铃木耦合反应(一般情况下,加热搅拌的温度可以为70~80℃,时间可以为40~55h,在实施例1中为75℃和48h);
48h后,反应液用二氯甲烷萃取,萃取后产物用饱和食盐水洗涤,再用旋转蒸发仪除去萃取剂;再对产物,以正己烷与二氯甲烷体积比为6∶1的溶液作为淋洗剂,进行硅胶层析柱分离提纯,收集产物,再用旋转蒸发仪除去溶剂,得到白色晶体产物4-(2-噻吩基)三苯胺c3。c3的分子式为c22h17ns,产率为72.0%。
s3.将0.0015mol中间产物c3加入经过遮光处理过的单口烧瓶中,加入40ml三氯甲烷,溶解反应物,开启搅拌器,并保持0℃冷浴的状态;15分钟后,少量多次地在30min内加入nbs(一般情况下,nbs可以为0.00153~0.00161mol,在实施例1中为0.0016mol),添加完毕后,持续搅拌并且冷浴(一般情况下,冷浴时间可以为6~7h,在实施例1中为6h);
6h后,利用旋转蒸发仪除去粗产品中的氯仿,用正己烷作为淋洗剂,进行硅胶层析柱分离提纯,收集产物,再用旋转蒸发仪除去淋洗剂,得到0.5350g淡黄色晶体4-(2-噻吩基)三苯胺衍生物c4。c4的名称为4-(2-溴-5-噻吩基)三苯胺,分子式为c22h16brns,产率为85.19%。
上述4-(2-噻吩基)三苯胺及其衍生物的合成线路如下:
实施例2
一种4-(2-噻吩基)三苯胺及其衍生物,其制备方法与实施例1的制备方法相比,区别在于,实施例2中的卤化试剂为n-碘代丁二酰亚胺,步骤s1.中卤化反应的反应条件为搅拌下-3℃冷浴8h;
其他原料用量及操作步骤与实施例1一致。
实施例3
一种4-(2-噻吩基)三苯胺及其衍生物,其制备方法与实施例1的制备方法相比,区别在于,实施例3的步骤s2.中suzuki偶联反应的温度为70℃,时间为48h;
其他原料用量及操作步骤与实施例1一致。
实施例4
一种4-(2-噻吩基)三苯胺及其衍生物,其制备方法与实施例1的制备方法相比,区别在于,实施例3的步骤s3.中卤化反应的反应条件为搅拌下-3℃冷浴6h;
其他原料用量及操作步骤与实施例1一致。
表征测试及结果
对实施例1制备得到的4-(2-噻吩基)三苯胺及其衍生物,即4-(2-噻吩基)三苯胺c3和4-(2-噻吩基)三苯胺衍生物c4进行如下结构表征和测试:
(1)核磁共振氢谱
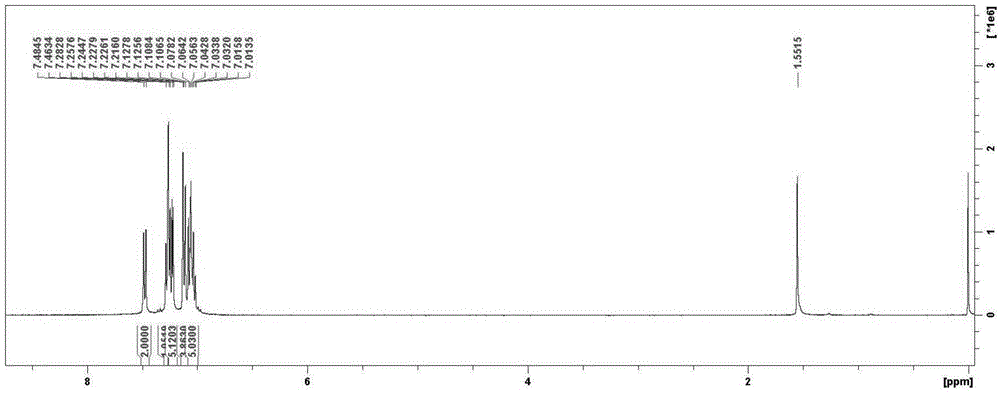
图1和图2分别为实施例1中c3和c4的核磁共振氢谱图。
c3的具体核磁数据为:1hnmr(400mhz,cdcl3),δ(ppm)7.485-7.463(d,j=8.5hz,2h),7.283-7.258(m,1h),7.225-7.216(m,5h),7.128-7.107(d,j=8.5hz,4h),7.078–7.014(m,5h)。
c4的具体核磁数据为:1hnmr(400mhz,cdcl3),δ(ppm)7.381-7.337(m,2h),7.287-7.248(m,4h),7.118-6.951(m,10h)。
(2)质谱
c3理论摩尔质量:327.11g/mol,ms(esi-qqq)found:m/z=327.95[m+h]+。
c4理论摩尔质量:405.22g/mol,ms(esi-qqq)found:m/z=405.89,407.86[m]+;406.88,408.77[m+h]+。
图3和图4分别为实施例1中c3和c4的核磁共振氢谱图,结果与实施例1制备的c3和c4的理论摩尔质量相一致。
(3)红外光谱
图5和图6分别为实施例1中c3和c4的红外光谱图。
对于c3:3030cm-1处的吸收峰是苯环中不饱和的c-h伸缩振动,1587-1482cm-1之间的峰为苯环的骨架振动,1328cm-1处为c-n伸缩振动,1587-1414cm-1应该归属于噻吩环上c=c对称伸缩振动及c=c不对称伸缩振动,1274cm-1处峰对应的是噻吩环上c-h的面内弯曲振动,817cm-1附近的峰应该是由噻吩环上c-h的面外弯曲振动引起,890-692cm-1之间的峰为芳环上c-h的面外变形振动,753cm-1和692cm-1处峰为芳环单取代,849-817cm-1之间峰为苯环对位二取代。
对于c4:3056cm-1附近的吸收峰是苯环中不饱和的c-h伸缩振动,1589-1486cm-1之间的峰为苯环的骨架振动,1317cm-1处为c-n伸缩振动,1589-1434cm-1应该归属于噻吩环上c=c对称伸缩振动及c=c不对称伸缩振动,1264cm-1处峰对应的是噻吩环上c-h的面内弯曲振动,838cm-1附近的峰应该是由噻吩环上c-h的面外弯曲振动引起,899-698cm-1之间的峰为芳环上c-h的面外变形振动,755cm-1和698cm-1处峰为芳环单取代,838-824cm-1之间峰为苯环对位二取代,525cm-1为c-br伸缩振动。
(4)紫外可见光吸收光谱
图7为实施例1中c3和c4的二氯甲烷溶液的紫外可见光吸收光谱图。将一定质量的c3和c4分别溶解于二氯甲烷溶液中,均配成溶度约为10-5mol/l的溶液,在室温下,测定c3和c4的紫外可见吸收光谱,归一化处理后如图7所示。c3和c4的峰形相似度极高,最大吸收峰出现在260~400nm之间。在引入溴原子后的c4,与c3相对比,发生了细微的红移。因为溴原子上的n电子,使得引起电子跃迁所需要的能量降低,所以吸收峰的波长会向长波长方向移动,而又因为三苯胺噻吩衍生物的共轭体系相对强大,所以溴原子只产生较小的影响(只发生细微的红移)。c4在二氯甲烷溶液中最大吸收波长为352nm,肩峰为238nm,低能量的吸收峰352nm对应于化合物c4的π-π*跃迁的吸收,因为二氯甲烷溶液的最低吸收波长为233nm,所以233nm附近的峰是由于二氯甲烷溶剂自身的影响而造成的,而较高能量的吸收峰238nm属于ch2cl2的n-π*跃迁。
图8为实施例1中c3和c4的薄膜的紫外可见光吸收光谱图。将少量的c3和c4分别溶解于二氯甲烷溶液中,并将溶液均匀涂抹于石英玻璃片上,干燥,即制得c3和c4薄膜。在室温下,测定c3和c4的薄膜紫外可见吸收光谱,其处理后的紫外可见光吸收光谱如图10所示。c3薄膜的紫外光吸收特征峰并不突出,而引入溴原子修饰后的c4,出现比较明显的紫外光吸收特征峰,在260~400nm之间出现了强吸收峰,这些吸收峰应该来自化合物c4自身π-π*的电子跃迁。
根据紫外可见吸收光谱图的结果,可以说明4-(2-噻吩基)三苯胺及其衍生物对紫外光有着良好的吸收能力,特别是对波长在260~400nm之间的紫外光。
(5)荧光光谱
以二氯甲烷为溶剂,将c3和c4分别配制成浓度约为10-5mol/l的溶液和薄膜。在室温下,用300nm的紫外光为激发波长,得到c3与c4的荧光发射光谱图。图9为实施例1中c3和c4的二氯甲烷溶液的荧光光谱图。图10为实施例1中c3和c4的薄膜的荧光光谱图。
如图9所示,c3的二氯甲烷溶液有着非常强烈的紫色荧光发射,最大发射峰位于420nm;而对比引入溴原子后的c4,c4的荧光发射强度剧烈下降,在相同的条件下,c4的发光强度不及c3的1%,基本可以认为c4不荧光发射,也验证了溴原子的重原子效应,能有效的猝灭化合物的荧光发射性能。
对图10进行分析可知,在薄膜状态下,c3最大发射峰出现在383nm附近,在375~425nm间有着比较明显的荧光发射,可以推断出c3的荧光颜色依然为紫色;而c4的荧光发射强度明显低很多,可以近似认为c4没有荧光发射,说明了引入溴原子后,可以猝灭化合物的荧光性能。
同样地,对实施例2~4制得的4-(2-噻吩基)三苯胺及其衍生物进行上述结构表征和测试。测试结果表明:实施例2~4制得的4-(2-噻吩基)三苯胺及其衍生物在紫外区内也具有很强的紫外吸收能力,并且式(ii)的4-(2-噻吩基)三苯胺及其衍生物不发荧光。
本发明以三苯胺为母体,通过卤化反应及suzuki偶联反应合成了4-(2-噻吩基)三苯胺及其衍生物,利用紫外可见光吸收光谱、分子荧光光谱等对产物进行性质分析,结果表明:4-(2-噻吩基)三苯胺及其衍生物对紫外线具有很强的吸收能力;相对于4-(2-噻吩基)三苯胺,当引入卤原子后,4-(2-噻吩基)三苯胺衍生物的紫外线吸收强度和范围(260~400nm)保持不变,而荧光发射强度大大下降,符合预期。
由此可知,本发明制备的4-(2-噻吩基)三苯胺及其衍生物对紫外区260~400nm范围内的紫外吸收能力较强,可以作为一种新型的紫外线吸收剂;此外,卤原子的重原子效应能产生荧光猝灭,使得4-(2-噻吩基)三苯胺衍生物不发荧光,不会对透过光线有影响,能够使原来的光线保持本色,同时也减小了对人眼睛的伤害。
显然,本发明的上述实施例仅仅是为清楚地说明本发明所作的举例,而并非是对本发明的实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明权利要求的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!