一种MEF2D1-97载体及其构建方法和应用与流程
本发明属于基因工程技术领域,具体涉及一种mef2d1-97载体及其构建方法和应用。
背景技术:
mef2d蛋白属于肌细胞特异性增强因子家族(mef2)成员,该家族成员包括mef2a、mef2b、mef2c、mef2d,均为具有重要生物学功能的转录因子。早期研究表明,mef2蛋白参与肌肉和神经细胞的分化及发育,与急性白血病的发病也有相关性,mef2蛋白还参与多个涉及细胞存活及凋亡的信号通路,如mapk-p38通路、cdk5、erk5,ampk及caspase通路等。对人类的癌症研究发现,mef2d在人肝癌中高表达,并促进肝癌细胞的增殖,mef2d被敲减后肝癌细胞的增殖水平降低,细胞阻滞在g2/m期;mef2d在肺癌中表达量也很高,降低其表达量也会影响肺癌细胞的增殖、存活及侵袭;胰腺癌中mef2d的表达也远高于癌旁组织,且mef2d的表达量与肿瘤的大小、组织分化程度及所处肿瘤分期阶段具有显著相关性,胰腺癌细胞系中敲除mef2d,会抑制细胞的增殖、迁移及侵袭能力,mef2d表达量降低也会减少磷酸化akt和gsk-3β的水平。mef2d表达与cd31阳性的结直肠癌组织中微血管密度具有正相关性,mef2d会诱导结直肠癌细胞中促血管新生因子的表达,促进肿瘤中血管生成;很多mirna也通过靶向调节mef2d对癌症细胞的功能进行调控,比如mir-30a通过降低mef2d的mrna及蛋白表达,抑制宫颈癌细胞的增殖和侵袭,mir-421通过抑制mef2d的表达抑制恶性胶质瘤的糖代谢、血管生成等功能。众多的研究结果提示,mef2d对多种癌症的发生发展以及癌细胞的生物学功能,具有重要作用。
双特异性酪氨酸磷酸化调节激酶1a(dyrk1a)基因,位于人类21号染色体的唐氏综合征关键区域(downsyndromecriticalregion,dscr),属于双特异性酪氨酸磷酸化调节激酶家族,是果蝇mnb基因(小脑基因)在哺乳动物中的同源基因。dyrk家族蛋白在调控细胞增殖的核功能的信号通路中起重要作用。dyrk1a可以催化自体丝、苏氨酸和酪氨酸磷酸化,不仅参与大脑的发育,在调控细胞增殖的细胞通路中也具有重要作用,是唐氏综合征(ds)的重要候选基因之一。dyrk1a的表达量及活性对细胞功能至为重要。dyrk1a通过发挥其激酶活性,磷酸化下游底物而参与细胞功能调控。因此dyrk1a的活性检测,意义重大。
目前的研究多是通过检测dyrk1a的自体酪氨酸磷酸化水平指示其激酶活性,应用免疫沉淀技术结合westernblot(wb)技术,通过与对照相比磷酸化条带的强弱水平,确定dyrk1a激酶活性的大小。该方法的弊端在于,操作步骤较为繁琐复杂,细胞裂解时要在裂解液中及时添加足量的仍处于效期内的蛋白酶抑制剂和磷酸酶抑制剂已减缓蛋白的降解以及磷酸化蛋白的去磷酸化速率,从收集细胞、细胞裂解、离心至免疫沉淀反应等需要全程保持在0-4℃条件下操作,且反应用时长,通常需要四小时至过夜孵育,长时间暴露于4℃环境即使在有蛋白酶抑制剂的条件下仍易导致蛋白降解。且免疫沉淀实验需要大量的裂解液,也需要琼脂糖珠,磷酸化抗体作为蛋白修饰抗体,与同规格普通蛋白抗体相比具有价格高昂,活性不够稳定、存放时间短等缺点,对结果的准确性有一定的影响。
其次,还可以通过检测dyrk1a的底物磷酸化水平来间接的反映dyrk1a的激酶活性。同样需要经过裂解细胞,蛋白定量并结合wb等方法,通过蛋白磷酸化抗体或者直接通过放射自显影来检测dyrk1a底物的磷酸化水平。蛋白磷酸化抗体依然存在以上描述的弊端,而放射自显影更需要放射性标记的32p,对检测者存在一定的射线危害。
中国专利cn104849448b公开了一种基于荧光淬灭的蛋白激酶活性分析方法,该方法是将靶标蛋白激酶与其对应的荧光标记多肽物混合,通过监测体系中荧光信号的淬灭情况来判定激酶活性,虽然具有放射性低、快速灵敏等优点,但同时该方法中使用到的纳米二氧化锫、荧光标记物等价格较高,对实验条件要求较高,容易对环境造成污染,且操作方法相对复杂。
总体来说,现有方法具有检测起来费时费力,结果不稳定也不够直观,且检测成本较高等缺点。
技术实现要素:
本发明的目的是针对现有技术中存在的缺点,提供一种mef2d1-97载体及其构建方法和应用。本发明提供的mef2d1-97载体,可以作为dyrk1a蛋白活性指示剂,具有灵敏度高,检测成本低,结果稳定,环境污染性低等优点。
为了达到上述目的,本发明所采用的技术方案为:
本发明提供了一种mef2d1-97载体,所述mef2d1-97载体是将mef2d1-97片段经人工合成后,插入到pcmv6-entry中构建而成的。
优选地,所述mef2d1-97片段为编码基因mef2d的n端前1-97个氨基酸的dna序列,具体序列信息如seqidno.1所示。
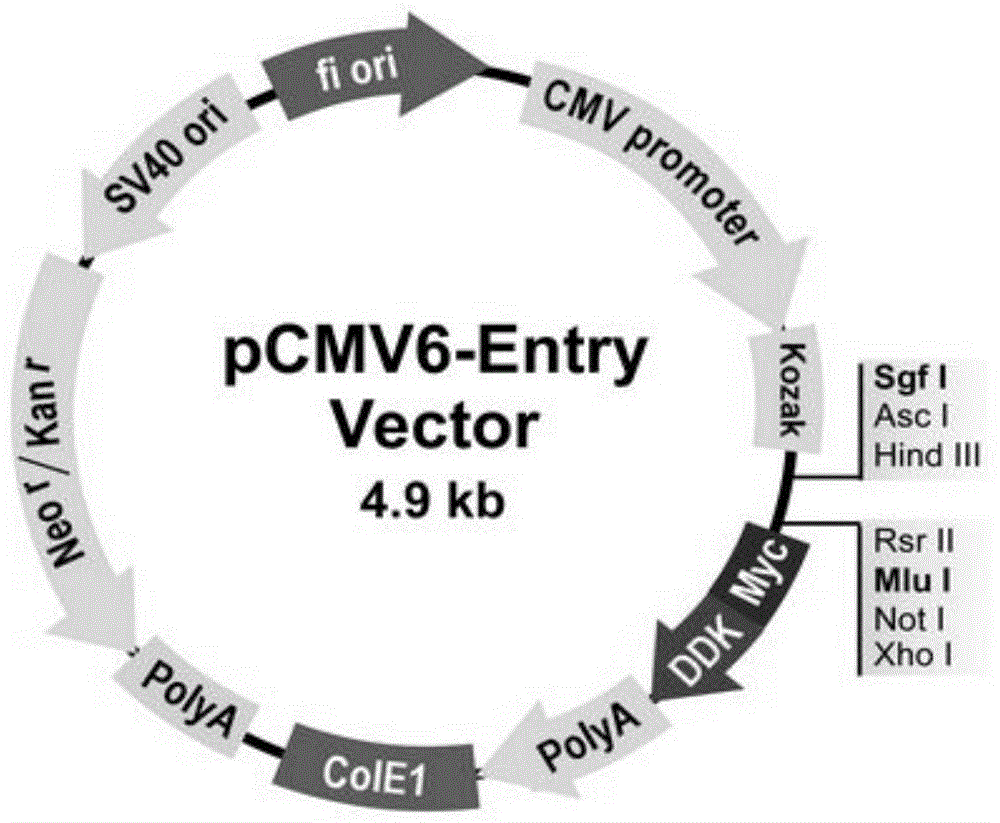
优选地,pcmv6-entry载体由origene公司提供,所述pcmv6-entry-mef2d载体是通过将mef2d的表达序列插入到原始载体pcmv6-entry(图1)的多克隆位点sgfi和mlui中,得到的mef2d与下游标签的融合蛋白序列。
本发明还提供了一种mef2d1-97表达载体的构建方法,包括以下步骤:
a.设计扩增mef2d蛋白的n端1-97个氨基酸序列的引物,并在引物上加入酶切位点,上游酶切位点为sgfi,下游酶切位点为mlui,以pcmv6-entry载体为模板,经pcr反应,得mef2d的n端1-97片段的pcr产物;
所述mef2d的n端1-97片段的dna序列为:
atggggaggaaaaagattcagatccagcgaatcaccgacgagcggaaccgacaggtgactttcaccaagcggaagtttggcctgatgaagaaggcgtatgagctgagcgtgctatgtgactgcgagatcgcactcatcatcttcaaccactccaacaagctgttccagtacgccagcaccgacatggacaaggtgctgctcaagtacacggagtacaatgagccacacgagagccgcaccaacgccgacatcatcgagaccctgaggaagaagggcttcaacggctgcgac;(seqidno.1)
b.将步骤a所得mef2d的n端1-97片段的pcr产物,取50-100μl进行琼脂糖凝胶电泳,并进行核酸纯化,得纯化产物;
c.将步骤b所得纯化产物与pcmv6-entry载体分别进行双酶切反应,后进一步纯化,得纯化的载体和插入片段;
d.将步骤c所得纯化的载体和插入片段做连接反应,用t4dna连接酶在16℃条件下连接1-3小时,得连接产物;
e.将步骤d所得连接产物转化入dh5α感受态细菌,并于转化后15-18小时后观察克隆生长情况,得单克隆;
f.挑取步骤e所得单克隆,在37℃条件下,于含有卡那霉素抗性的lb培养基中摇菌15-20小时,并纯化菌体质粒,进行酶切验证,得酶切正确的质粒;
g.将步骤f所得酶切正确的纯化质粒送公司测序,挑选测序结果与mef2d1-97原序列一致的质粒,即得。
优选地,所述步骤a中扩增mef2d蛋白的n端1-97个氨基酸的引物为a,其dna序列为:
f:gccgcgatcgccatggggaggaaaaagattc
r:cgtacgcgtgtcgcagccgttgaagccc
优选地,所述步骤a中pcr反应时的扩增程序为:94℃3min;94℃30s,55℃30s,72℃30s,35个循环;72℃10min。
优选地,所述步骤c中的双酶切反应时,所用限制性内切酶为sgfi和mlui,反应条件:37℃条件下酶切15-20分钟。
本发明利用westernblot检测载体的表达是否正确,操作步骤如下:
1)应用pei转染试剂将构建好的载体分别转染入hek293细胞中,hek293细胞转染时密度约为70-80%,转染时间为40-60小时,得到表达mef2d1-97蛋白的hek293细胞;
2)于4℃,12000rpm的条件下离心收取步骤1)所得含mef2d1-97蛋白的hek293细胞,向细胞沉淀中加入含有蛋白酶抑制剂的裂解液,超声裂解后于4℃,12000rpm的条件下离心10-20分钟,留取上清,得蛋白样品;
3)将步骤2)所得的蛋白样品利用bca法测定蛋白质浓度,每个样品取50ug总蛋白量,加入上样缓冲液混匀并于95℃加热变性5min,冷却至室温,将样品进行wb电泳,应用anti-flag一抗结合对应种属的二抗进行检测。
本发明还提供了一种所述的mef2d1-97的载体在检测dyrk1a蛋白活性中的应用。
本发明所述mef2d1-97的载体在检测dyrk1a蛋白活性中的应用,是利用westernblot检测dyrk1a诱导的mef2d1-97表达的改变情况,操作步骤如下:
1)应用pei转染试剂将构建好的pcmv6-entry-mef2d1-97质粒分别与dyrk1a表达质粒及空白对照质粒共转染入60mm培养皿的hek293细胞中,转染时细胞密度约为70-80%,转染4-5小时后更换培养基;
2)共转染40-56小时后,弃去培养基并于4℃,12000rpm离心5-10min,收集细胞,用预冷的pbs清洗细胞,并于相同条件下再次离心收集,向细胞沉淀中加入含足量蛋白酶抑制剂及磷酸酶抑制剂的ripa裂解液300μl,在30%的强度下超声充分破碎细胞,4℃条件下12000rpm离心10-20min后,取上清液,利用bca法进行蛋白定量;
3)根据定量结果将每个样品总蛋白取出50μg,按样品与6×sds上样变性缓冲液体积比为5:1的比例混匀,制备混悬液,95℃加热5分钟后冷却离心,将样品上样于12%glycinesds-page凝胶电泳,进行wb检测,应用anti-flag一抗结合对应种属的二抗,观察mef2d1-97表达的差异。
优选地,所述mef2d1-97的载体在检测dyrk1a蛋白活性中的应用时,还可以通过构建mef2d1-97-gfp载体,然后利用免疫荧光技术检测dyrk1a诱导的mef2d1-97表达的改变情况,具体操作步骤为:
1)将mef2d1-97的序列与egep的蛋白序列融合,并去掉egfp的起始密码子atg,使两者之间不间隔任何序列,将融合序列直接进行合成,得融合插入序列;
2)将步骤1)所得融合插入序列,克隆至penter载体,克隆的双酶切位点分别为sgfi和mlui,经连接,转化,验证,测序,即得mef2d1-97-gfp载体;
3)将步骤2)所得mef2d1-97-gfp表达质粒与dyrk1a表达质粒及空白对照质粒分别共转染入35mm培养皿的hek293细胞中,转染24-48小时后,进行活细胞的荧光拍照及定量分析。
优选地,所述dyrk1a表达载体是通过将dyrk1a的表达序列插入到原始载体pcmv6-entry(图2)的多克隆位点sgfi和mlui中,得到的dyrk1a与下游标签的融合蛋白序列。
优选地,所述ripa裂解液包括包括50mmtris,其ph值为7.4,150mmnacl,1%tritonx-100,1%sodiumdeoxycholate,0.1%sds。
优选地,所述6×sds上样变性缓冲液包括ph值为6.8的4×tris-hcl7ml;甘油3ml;sds1g;二硫苏糖醇0.93g或β-巯基乙醇6ml;溴酚蓝1.2mg;纯化水定容至10ml。
优选地,所述pei转染试剂购自山东维真生物科技有限公司;所述mef2d表达载体及dyrk1a表达载体购自origene公司;所述蛋白酶抑制剂、磷酸酶抑制剂及anti-flag抗体购自sigma公司;所述蛋白定量检测试剂盒购自美国的bio-rad公司;所述培养基为含10%胎牛血清的高糖dmem培养基,其中,10%的胎牛血清购自thermofisher公司,培养基购自北京中科迈晨科技有限公司。
与现有技术相比,本发明提供的mef2d1-97载体具有以下优势:
(1)本发明提供的mef2d1-97载体,构建过程中采用标签抗体取代传统的磷酸化抗体,标签抗体更易于获得及保存,稳定性更好,降低了检测的成本;
(2)本发明提供的mef2d1-97载体,可以作为dyrk1a蛋白活性指示剂检测dyrk1a蛋白活性,具有检测效果更为直观,检测过程更为方便灵敏;
(3)本发明提供的mef2d1-97载体,利用westernblot检测构建表达载体的准确性,去掉了免疫沉淀的步骤,细胞直接裂解定量后上样,避免了操作步骤繁琐导致的出错率和样品损失,同时减少蛋白暴露在4℃环境中的时间,降低蛋白降解,检测结果更为准确;
(4)本发明提供的mef2d1-97-gfp载体检测dyrk1a蛋白活性时,可以利用免疫荧光技术,免疫荧光法检测相对于wb检测,效果更为直观,检测更为方便灵敏,检测时间短,同时对wb的检测结果进行双重验证。
附图说明
图1为pcmv6-entry载体多克隆位点图谱;
图2为penter载体多克隆位点图谱;
图3为实施例3中利用wb技术检测dyrk1a对mef2d表达的调节;
图4为实施例3中经wb检测dyrk1a对mef2d1-97的表达量的影响;
图5为实施例3中经免疫共沉淀免疫荧光实验验证mef2d和dyrk1a在hek293细胞中的特异性结合情况;
图6为经免疫荧光实验验证mef2d和dyrk1a在hek293细胞中的特异性结合情况;
图7为利用荧光显微镜观察到的mef2d1-97本身的表达量图;
图8为利用荧光显微镜观察到的在dyrk1a活性高的时候,mef2d1-97的表达量图。
具体实施方式
为了更好的说明本发明的目的、技术方案和优点,下面将结合具体实施例对本发明作进一步说明。
实施例1一种mef2d1-97载体
所述mef2d1-97载体,是将mef2d1-97片段经人工合成后,插入到pcmv6-entry中构建而成的。
所述mef2d1-97表达载体的构建方法为:
a.设计扩增mef2d蛋白的n端1-97个氨基酸序列的引物,引物序列见表1,并在引物上加入酶切位点,上游酶切位点为sgfi,下游酶切位点为mlui,以pcmv6-entry载体为模板,进行pcr反应,pcr反应程序为94℃3min;94℃30s,55℃30s,72℃30s,35个循环;72℃10min,反应结束后,得mef2d的n端1-97片段;
b.将步骤a所得mef2d的n端1-97片段,取50μl进行琼脂糖凝胶电泳,并进行核酸纯化,得纯化产物;
c.将步骤b所得纯化产物与pcmv6-entry载体分别进行双酶切反应,所用限制性内切酶为sgfi和mlui,反应条件为37℃条件下酶切15分钟后进一步纯化,得纯化的载体和插入片段;
d.将步骤c所得纯化的载体和插入片段做连接反应,用t4dna连接酶在16℃条件下连接1小时,得连接产物;
e.将步骤d所得连接产物转化入dh5α感受态细菌,并于转化后15小时后观察克隆生长情况。得单克隆;
f.挑取步骤e所得单克隆,在37℃条件下,于含有卡那霉素抗性的lb培养基中摇菌15小时,并纯化菌体质粒,进行酶切验证。得酶切正确的质粒;
g.将步骤f所得酶切正确的纯化质粒送公司测序,挑选测序结果与mef2d1-97原序列一致的质粒,即得。
表1扩增mef2d1-97个片段的引物序列
所述载体的表达是否正确的检测方法为:
1)应用pei转染试剂将构建好的载体分别转染入hek293细胞中,hek293细胞转染时密度约为70%,转染时间为40小时,得到表达mef2d1-97蛋白的hek293细胞;
2)于4℃,12000rpm的条件下离心收取步骤1)所得含mef2d1-97蛋白的hek293细胞,向细胞沉淀中加入含有蛋白酶抑制剂的裂解液,超声裂解后于4℃,12000rpm的条件下离心10分钟,留取上清,得蛋白样品;
3)将步骤2)所得的蛋白样品利用bca法测定蛋白质浓度,每个样品取50μg总蛋白量,加入上样缓冲液混匀并于95℃加热变性5min,冷却至室温,将样品进行wb电泳,应用anti-flag一抗结合对应种属的二抗进行检测。
实施例2一种mef2d1-97载体
所述mef2d1-97载体,是将mef2d1-97片段经人工合成后,插入到pcmv6-entry-mef2d中构建而成的。
所述mef2d1-97表达载体的构建方法为:
a.设计扩增mef2d蛋白的n端1-97个氨基酸序列的引物,具体的引物序列见表1,并在引物上加入酶切位点,上游酶切位点为sgfi,下游酶切位点为mlui,以pcmv6-entry载体为模板,进行pcr反应,pcr反应程序为94℃3min;94℃30s,55℃30s,72℃30s,35个循环;72℃10min,反应结束后,得mef2d的n端1-97片段;
b.将步骤a所得mef2d的n端1-97片段,取100μl进行琼脂糖凝胶电泳,并进行核酸纯化,得纯化产物;
c.将步骤b所得纯化产物与pcmv6-entry载体分别进行双酶切反应,所用限制性内切酶为sgfi和mlui,反应条件为37℃条件下酶切20分钟后进一步纯化,得纯化的载体和插入片段;
d.将步骤c所得纯化的载体和插入片段做连接反应,用t4dna连接酶在16℃条件下连接3小时,得连接产物;
e.将步骤d所得连接产物转化入dh5α感受态细菌,并于转化后18小时后观察克隆生长情况。得单克隆;
f.挑取步骤e所得单克隆,在37℃条件下,于含有卡那霉素抗性的lb培养基中摇菌20小时,并纯化菌体质粒,进行酶切验证。得酶切正确的质粒;
g.将步骤f所得酶切正确的纯化质粒送公司测序,挑选测序结果与mef2d1-97原序列一致的质粒,即得。
所述载体的表达是否正确的检测方法为:
1)应用pei转染试剂将构建好的载体分别转染入hek293细胞中,hek293细胞转染时密度约为80%,转染时间为60小时,得到表达mef2d1-97蛋白的hek293细胞;
2)于4℃,12000rpm的条件下离心收取步骤1)所得含mef2d1-97蛋白的hek293细胞,向细胞沉淀中加入含有蛋白酶抑制剂的裂解液,超声裂解后于4℃,12000rpm的条件下离心20分钟,留取上清,得蛋白样品;
3)将步骤2)所得的蛋白样品利用bca法测定蛋白质浓度,每个样品取50μg总蛋白量,加入上样缓冲液混匀并于95℃加热变性5min,冷却至室温,将样品进行wb电泳,应用anti-flag一抗结合对应种属的二抗进行检测。
实施例3一种mef2d1-97载体
所述mef2d1-97载体,是将mef2d1-97片段经人工合成后,插入到pcmv6-entry-mef2d中构建而成的。
所述mef2d1-97表达载体的构建方法为:
a.设计扩增mef2d蛋白的n端1-97个氨基酸序列的引物,引物序列见表1,并在引物上加入酶切位点,上游酶切位点为sgfi,下游酶切位点为mlui,以pcmv6-entry-mef2d载体为模板,进行pcr反应,pcr反应程序为94℃3min;94℃30s,55℃30s,72℃30s,35个循环;72℃10min,反应结束后,得mef2d的n端1-97片段;
b.将步骤a所得mef2d的n端1-97片段,取75μl进行琼脂糖凝胶电泳,并进行核酸纯化,得纯化产物;
c.将步骤b所得纯化产物与pcmv6-entry载体分别进行双酶切反应,所用限制性内切酶为sgfi和mlui,反应条件为37℃条件下酶切18分钟后进一步纯化,得纯化的载体和插入片段;
d.将步骤c所得纯化的载体和插入片段做连接反应,用dna连接酶在16℃条件下连接3小时,得连接产物;
e.将步骤d所得连接产物转化入dh5α感受态细菌,并于转化后18小时后观察克隆生长情况,得单克隆;
f.挑取步骤e所得单克隆,在37℃条件下,于含有卡那霉素抗性的lb培养基中摇菌18小时,并纯化菌体质粒,进行酶切验证,得酶切正确的质粒;
g.将步骤f所得酶切正确的纯化质粒送公司测序,挑选测序结果与mef2d1-97原序列一致的质粒,即得。
所述载体的表达是否正确的检测方法为:
1)应用pei转染试剂将构建好的载体分别转染入hek293细胞中,hek293细胞转染时密度约为75%,转染时间为50小时,得到表达mef2d1-97蛋白的hek293细胞;
2)于4℃,12000rpm的条件下离心收取步骤1)所得含mef2d1-97蛋白的hek293细胞,向细胞沉淀中加入含有蛋白酶抑制剂的裂解液,超声裂解后于4℃,12000rpm的条件下离心15分钟,留取上清,得蛋白样品;
3)将步骤2)所得的蛋白样品利用bca法测定蛋白质浓度,每个样品取50μg总蛋白量,加入上样缓冲液混匀并于95℃加热变性5min,冷却至室温,将样品进行wb电泳,应用anti-flag一抗结合对应种属的二抗进行检测。
实施例4一种检测dyrk1a诱导mef2d1-97表达改变的方法
所述dyrk1a诱导mef2d1-97表达的改变情况,采用westernblot技术,具体操作步骤为:
1)应用pei转染试剂将实施例3构建好的pcmv6-entry-mef2d1-97质粒分别与dyrk1a表达质粒及空白对照质粒共转染入60mm培养皿的hek293细胞中,转染时细胞密度约为70%,转染4小时后更换培养基;
2)共转染40小时后,弃去培养基并于4℃,12000rpm离心5min,收集细胞,用预冷的pbs清洗细胞,并于相同条件下再次离心收集,向细胞沉淀中加入含足量蛋白酶抑制剂及磷酸酶抑制剂的ripa裂解液300μl,在30%的强度下超声充分破碎细胞,4℃条件下12000rpm离心10min后,取上清液,利用bca法进行蛋白定量;
3)根据定量结果将每个样品总蛋白取出50μg,按样品与6×sds上样变性缓冲液体积比为5:1的比例混匀,95℃加热5分钟后冷却离心,将样品上样于12%glycinesds-page凝胶电泳,进行wb检测,应用anti-flag一抗结合对应种属的二抗,观察mef2d1-97表达的差异。
实施例5一种检测dyrk1a诱导mef2d1-97表达改变的方法
所述dyrk1a诱导mef2d1-97表达的改变情况,采用westernblot技术,具体操作步骤为:
1)应用pei转染试剂将实施例3构建好的pcmv6-entry-mef2d1-97质粒分别与dyrk1a表达质粒及空白对照质粒共转染入60mm培养皿的hek293细胞中,转染时细胞密度约为80%,转染5小时后更换培养基;
2)共转染56小时后,弃去培养基并于4℃,12000rpm离心10min,收集细胞,用预冷的pbs清洗细胞,并于相同条件下再次离心收集,向细胞沉淀中加入含足量蛋白酶抑制剂及磷酸酶抑制剂的ripa裂解液300μl,在30%的强度下超声充分破碎细胞,4℃条件下12000rpm离心20min后,取上清液,利用bca法进行蛋白定量;
3)根据定量结果将每个样品总蛋白取出50μg,按样品与6×sds上样变性缓冲液体积比为5:1的比例混匀,95℃加热5分钟后冷却离心,将样品上样于12%glycinesds-page凝胶电泳,进行wb检测,应用anti-flag一抗结合对应种属的二抗,观察mef2d1-97表达的差异。
实施例6一种检测dyrk1a诱导mef2d1-97表达改变的方法
所述dyrk1a诱导mef2d1-97表达的改变情况,采用westernblot技术,具体操作步骤为:
1)应用pei转染试剂将实施例3构建好的pcmv6-entry-mef2d1-97质粒分别与dyrk1a表达质粒及空白对照质粒共转染入60mm培养皿的hek293细胞中,转染时细胞密度约为75%,转染4.5小时后更换培养基;
2)共转染48小时后,弃去培养基并于4℃,12000rpm离心8min,收集细胞,用预冷的pbs清洗细胞,并于相同条件下再次离心收集,向细胞沉淀中加入含足量蛋白酶抑制剂及磷酸酶抑制剂的ripa裂解液300μl,在30%的强度下超声充分破碎细胞,4℃条件下12000rpm离心15min后,取上清液,利用bca法进行蛋白定量;
3)根据定量结果将每个样品总蛋白取出50μg,按样品与6×sds上样变性缓冲液体积比为5:1的比例混匀,95℃加热5分钟后冷却离心,将样品上样于12%glycinesds-page凝胶电泳,进行wb检测,应用anti-flag一抗结合对应种属的二抗,观察mef2d1-97表达的差异。
利用westernblot技术检测dyrk1a对mef2d表达的调节,dyrk1a可以磷酸化mef2d并升高mef2d的蛋白表达,磷酸化后的mef2d可以被碱性磷酸酶逆转,说明蛋白磷酸化修饰的切实存在(图3),进一步检测mef2d1-97的表达情况,说明dyrk1a可以显著升高mef2d1-97的表达量(图4)。
实施例7一种检测dyrk1a诱导mef2d1-97表达改变的方法
所述dyrk1a诱导mef2d1-97表达的改变情况,采用免疫荧光技术,具体操作步骤如下:
1)将mef2d1-97的序列与egep的蛋白序列融合,并去掉egfp的起始密码子atg,使两者之间不间隔任何序列,将融合序列直接进行合成,得融合插入序列;
2)将步骤1)所得融合插入序列,克隆至penter载体,克隆的双酶切位点分别为sgfi和mlui,按照实施例3的方式进行连接,转化,验证,测序,即得mef2d1-97-gfp载体;
3)将步骤2)所得mef2d1-97-gfp表达质粒与dyrk1a表达质粒及空白对照质粒分别共转染入35mm培养皿的hek293细胞中,转染46小时后,进行活细胞的荧光拍照及定量分析。
利用免疫荧光技术结合免疫共沉淀技术,双重验证了mef2d和dyrk1a的相互作用(图5,图6),通过荧光显微镜的结果可以更直观的检测到mef2d1-97本身的表达量(图7)及与dyrk1a过量表达后,mef2d1-97的表达量显著上升(图8)。
最后应当说明的是,以上实施例仅用以说明本发明的技术方案而非对本发明保护范围的限制,尽管参照较佳实施例对本发明做了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的实质和范围。
序列表
<110>山东大学齐鲁医院
<120>一种mef2d1-97载体及其构建方法和应用
<130>2018.12.5
<160>1
<170>siposequencelisting1.0
<210>1
<211>291
<212>dna
<213>人工合成(artificialsynthesis)
<400>1
atggggaggaaaaagattcagatccagcgaatcaccgacgagcggaaccgacaggtgact60
ttcaccaagcggaagtttggcctgatgaagaaggcgtatgagctgagcgtgctatgtgac120
tgcgagatcgcactcatcatcttcaaccactccaacaagctgttccagtacgccagcacc180
gacatggacaaggtgctgctcaagtacacggagtacaatgagccacacgagagccgcacc240
aacgccgacatcatcgagaccctgaggaagaagggcttcaacggctgcgac291
- 还没有人留言评论。精彩留言会获得点赞!