一种基于高通量测序技术用于检测甲状腺癌致病相关基因变异的引物及其应用的制作方法
本发明涉及分子生物学技术领域,具体涉及一种基于高通量测序技术用于检测甲状腺癌致病相关基因变异的建库引物,建库方法及试剂盒。
背景技术:
经统计,触诊检查出甲状腺结节的发病率为5%,通过超声检查为35%。这些结节中85%~95%为良性结节,如结节性甲状腺肿和甲状腺腺瘤,仅有5%~15%的结节为恶性结节,即甲状腺癌。甲状腺癌诊断上,细针穿刺细胞学检查(fna)是当今评估甲状腺结节是否为恶性肿瘤最可靠的手段。然而fna无法明确诊断20%-30%的甲状腺结节,此种案例被报道为意义不明确。由于缺乏可靠的诊断手段,部分患者需通过术后病理检测明确诊断,造成患者不必要的治疗伤害。如果术中甲状腺叶切除术标本显示癌灶大于1cm,需要二次手术完成甲状腺全切术。可是如果对这类无法明确诊断的结节进行保守治疗,又使得恶性肿瘤患者存在淋巴结扩散和转移的巨大风险。在美国,甲状腺癌分子诊断作为一种准确性、敏感度高的诊断技术或辅助诊断技术已应用于临床,为医生诊断甲状腺结节的良恶性提供可靠信息,为个体化医疗提供依据。
有文献报道,甲状腺癌的遗传学改变包括:丝裂原活化蛋白激酶(mapk)通路及磷酸肌醇-3-激酶(pi3k)/akt信号通路。目前为止,超过90%的甲状腺癌基因变异已被阐述清楚。在这些非重叠的遗传学改变中,70%的甲状腺乳头状癌(ptcs)证实发现了braf、ras、ret/ptc癌基因;70%的甲状腺滤泡癌(ftc)证实发现了ras及pax8/pparc癌基因;低度分化及未分化癌中可见tp53癌基因。无论是家族性还是分散性的甲状腺髓样癌(mtc)在ret基因或者ras基因上常发生点突变。这些突变基因的标记、基因表达的改变已经在甲状腺结节的诊断中探讨了其作用,其中一些已在临床上得到应用。braf是研究最多的癌基因之一,也是甲状腺癌遗传学改变最常见的突变基因。
美国2017nccn临床实践指南详细指出对细针抽吸细胞学检查所见如下病理结果:1.疑似滤泡型及hurthle细胞癌(诊断依据为血管或包膜的侵犯,故不能用细针抽吸确诊)放射照像无法确定恶性利用分子诊断做进一步检测;2.意义不明的滤泡损伤放射照像无法确定恶性利用分子诊断做进一步检测;3.细胞学无法明确诊断的结节利用分子诊断判断良性/恶性。nccn临床实践指南中指出braf、ras、ret/ptc、pax8/pparg癌基因的变异检测对辅助诊断甲状腺癌具有重要作用。
目前针对二代测序技术中的靶向捕获技术主要包括多重pcr技术、靶向捕获技术。靶向捕获技术,适合大数据的靶向捕获,但开发成本高,操作复杂;多重pcr捕获技术操作简单,灵活,适合中小通量的数据量。
本发明为针对甲状腺癌相关基因的变异检测,包括braf、nras、kras、hras、tert、tp536个基因的18个突变位点和pax8/pparg、ret/ptc1(ret/ccdc6)和ret/ptc3(ret/ncoa4)3个融合基因,覆盖了90%~95%甲状腺癌致病基因变异。brafv600e突变与甲状腺乳头状癌密切相关,存在于70%~80%的甲状腺乳头状癌中。ras突变发生于15%~20%的甲状腺乳头状癌中。tert启动子突变于甲状腺癌的恶性程度及预后显著相关,是甲状腺癌诊断和治疗的新的分子标志物。tp53基因所编码的p53蛋白在维持细胞正常生长、抑制恶性增殖过程中起重要作用,在甲状腺癌中,认为tp53基因与甲状腺癌去分化有关,提示预后不良。ret/ptc基因重排,最常见的类型为位于10号染色体的长臂的ret/ptc1(ret/ccdc6)和ret/ptc3(ret/ncoa4)基因重排,由ret基因分别与h4基因和ele1基因臂内倒位融合形成,使ret/ptc蛋白质能激活mapk信号通路,乳头状甲状腺癌患者常发生ret/ptc基因重排,尤其在年轻患者和曾辐射暴露的患者中频发,在临床上,ret重排阳性的甲状腺癌预后相对较好。pax8/pparγ重排导致第2号和3号染色体发生易位,从而引起编码特异的甲状腺匹配区转录因子的pax8基因与pparγ基因发生融合。pax8/pparγ重排发生于常规的滤泡癌和嗜酸细胞癌,可能与肿瘤的演进、侵袭、转移相关。
技术实现要素:
本发明需要解决的技术问题是,提供一种特异性捕获甲状腺癌相关基因位点的序列的灵敏、准确、操作简便的高通量测序建库试剂盒,实现对临床甲状腺病变的检测与辅助诊断,以弥补现有检测方法的不足。
为解决上述技术问题,本发明采用以下技术方案:
一种基于高通量测序技术用于检测甲状腺癌致病相关基因变异的引物,包括seqidno:1~37所示的核苷酸序列。分别利用样本的dna和rna构建两个文库:利用样本dna构建检测braf、nras、kras、hras、tert、tp536个基因的18个突变位点的文库,利用样本rna逆转录成的cdna构建检测ccdc6/ret、ncoa4/ret、pax8/pparg3个融合基因的文库,其中扩增gapdh和krt7作为内参。
一种基于高通量测序技术用于检测甲状腺癌致病相关基因变异的试剂盒,包括seqidno:1~37所示的核苷酸序列。
检测甲状腺癌致病相关基因变异的方法,包括如下步骤:
(1)提取样本中的dna和rna;
(2)样本rna进行逆转录制备cdna;
(3)构建测序文库:
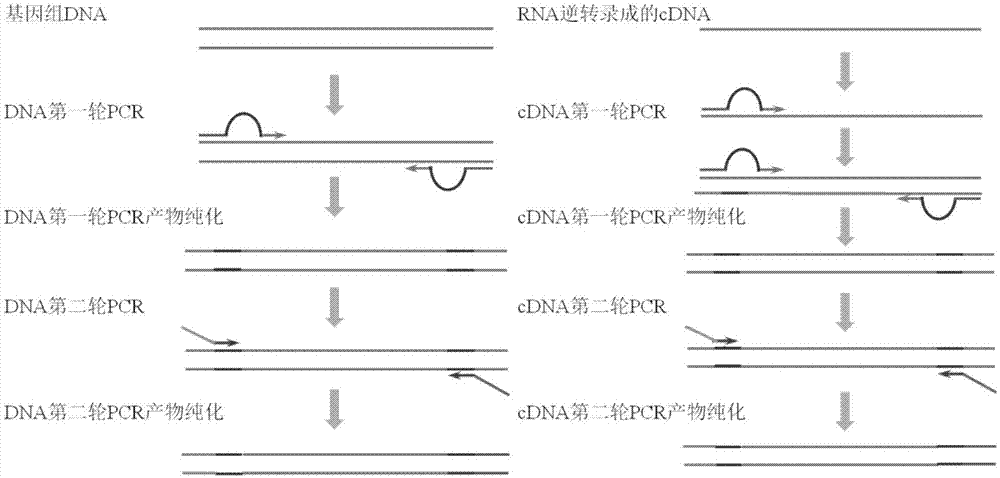
(a)以seqidno.1~22所示的核苷酸序列为引物,样本dna为模板,进行第一轮pcr扩增,纯化pcr产物,得到第一轮dnapcr产物;
(b)以第一轮dnapcr产物为模板,seqidno:23~24所示的序列为引物,进行第二轮pcr扩增,纯化pcr产物,得到第二轮dnapcr产物,得到dna测序文库;
(c)以seqidno:25~37所示的核苷酸序列为引物,步骤(2)得到的cdna为模板,进行第一轮pcr扩增,纯化pcr产物,得到第一轮rnapcr产物;
(d)以第一轮rnapcr产物为模板,seqidno:23~24所示的序列为引物,进行第二轮pcr扩增,纯化pcr产物,得到第二轮rnapcr产物,得到rna测序文库;
(4)上机测序;
(5)生物信息学分析软件对进行数据结果分析。
检测甲状腺癌致病相关基因变异的方法,具体如下:
样本dna构建检测braf、nras、kras、hras、tert、tp536个基因的18个突变位点的文库,包括如下步骤:
步骤1a,dna样本中加入第一轮扩增的dna特异性引物混合池和高保真pcr酶混合液制成pcr总体系,进行第一轮扩增;
步骤2a,第一轮pcr产物加入等体积的磁珠进行纯化;
步骤3a,纯化后的第一轮pcr产物,加入第二轮扩增的通用引物对和高保真pcr酶混合液制成pcr总体系,进行第二轮扩增;
步骤4a,第二轮pcr产物加入0.8倍体积的磁珠进行纯化,纯化后所得产物即可稀释到合适的浓度,用于二代测序。
其中,
所述步骤1a中,dna特异性引物混合池包括核苷酸序列seqidno:1~22所示的单链dna,dna特异性引物混合池浓度为10um;
所述步骤2a和4a中,所用磁珠为beckmanagencourtampurexp磁珠;
所述步骤3a中,通用引物对即为核苷酸序列seqidno:23~24所示的单链dna,通用引物浓度为10um。
利用样本rna逆转录成的cdna构建检测ccdc6/ret、ncoa4/ret、pax8/pparg3个融合基因的文库,其中扩增gapdh和krt7作为内参,包括如下步骤:
步骤1b,cdna样本中加入第一轮扩增的rna特异性引物混合池和高保真pcr酶混合液制成pcr总体系,进行第一轮扩增;
步骤2b,第一轮pcr产物加入等体积的磁珠进行纯化;
步骤3b,纯化后的第一轮pcr产物,加入第二轮扩增的通用引物对和高保真pcr酶混合液制成pcr总体系,进行第二轮扩增;
步骤4b,第二轮pcr产物使用0.8倍体积的磁珠纯化两遍,纯化后所得产物即可稀释到合适的浓度,用于二代测序。
其中,
所述步骤1b中rna特异性引物混合池包括核苷酸序列seqidno:25~37所示的单链dna,rna特异性引物混合池浓度为10um;
所述步骤2b和4b中所用磁珠为beckmanagencourtampurexp磁珠;
所述步骤3b中通用引物对即为核苷酸序列seqidno:23~24所示的单链dna,通用引物浓度为10um。
一种基于高通量测序技术用于检测甲状腺癌致病相关基因变异的试剂盒,检测的变异位点具体如下:
一种基于高通量测序技术用于检测甲状腺癌致病相关基因变异的试剂盒,该试剂盒包括如下试剂:
(1)高保真pcr酶混合液:该反应液中含有酶缓冲液、mg2+、dntps、高保真dna聚合酶;
(2)dna特异性引物:seqidno:1~23;
(3)rna特异性引物:seqidno:25~37;
(4)通用引物:seqidno:24~25;
(5)阴性对照:去rna酶的水。
甲状腺结节中大部分为良性结节,仅有5%~15%的结节为甲状腺癌。目前,手术切除是治疗甲状腺癌的主要方式,术前对结节的良恶性鉴别对于选择治疗方式、避免不必要的手术具有重要的作用,但是目前常见的临床诊断技术仍有20%-30%的甲状腺结节在术前无法明确诊断良恶性。本发明通过检测甲状腺癌致病相关基因变异来辅助诊断意义不明确的甲状腺结节病变。
有益效果:
(1)通过sci文献和数据库检索本试剂盒确定了以上九个基因的检测范围,覆盖了甲状腺癌90%~95%的变异位点;
(2)本发明为基于高通量测序平台的甲状腺癌致病相关基因变异的试剂盒具有通量高,灵敏度高,特异性强,准确度高的特点;
(3)本发明为多重pcr建库技术路线,通过两次pcr扩增即可完成建库具有建库时间短,操作简便的特点;
(4)本发明一种基于高通量测序技术用于检测甲状腺癌致病相关基因变异的试剂盒可用于辅助临床诊断意义不明确的甲状腺结节,判断甲状腺病变类型,为临床治疗提供参考依据。
附图说明
图1为本试剂盒两轮多重pcr的二代测序建库过程。
图2为本试剂盒构建的dna文库和cdna文库的片段分布图。
图3为实施例5临床样本检测20个样本18个snv位点的测序深度。
具体实施方式
根据下述实施例,可以更好地理解本发明。然而,本领域的技术人员容易理解,实施例所描述的内容仅用于说明本发明,而不应当也不会限制权利要求书中所详细描述的本发明。
实施例1:扩增甲状腺癌相关基因变异位点的核酸序列,由铂尚生物技术(上海)有限公司合成:
扩增braf的引物对:
braf_f1(seqidno.1):
5′-acacgacgctcttccgatctctttacttactacacctcaga-3′,
braf_r1(seqidno.2):
5′-gacgtgtgctcttccgatctgatgggacccactccatcgaga-3′;
扩增kras的引物对:
kras_f1(seqidno.3):
5′-acacgacgctcttccgatctctaatatagtcacattttcatt-3′,
kras_r1(seqidno.4):
5′-gacgtgtgctcttccgatctagctgtatcgtcaaggcactc-3′;
kras_f2(seqidno.5):
5′-acacgacgctcttccgatcttggtccctcattgcactgtactc-3′,
kras_r2(seqidno.6):
5′-gacgtgtgctcttccgatcttgtttctcccttctcaggattc-3′;
kras_f3(seqidno.7):
5′-acacgacgctcttccgatctagatctgtatttatttcagtg-3′,
kras_r3(seqidno.8):
5′-gacgtgtgctcttccgatctaacagtagacacaaaacaggct-3′;
扩增nras的引物对:
nras_f1(seqidno.9):
5′-acacgacgctcttccgatctactggtttccaacaggttctt-3′,
nras_r1(seqidno.10):
5′-gacgtgtgctcttccgatcttggttctggattagctggattg-3′;
nras_f2(seqidno.11):
5′-acacgacgctcttccgatctcacagaggaagccttcgcctg-3′,
nras_r2(seqidno.12):
5′-gacgtgtgctcttccgatctggttatagatggtgaaacctg-3′;
nras_f3(seqidno.13):
5′-acacgacgctcttccgatctacctatggtgctagtgggaaa-3′,
nras_r3(seqidno.14):
5′-gacgtgtgctcttccgatctgaaagctgtaccatacctgtct-3′;
扩增hras的引物对:
hras_f1(seqidno.15):
5′-acacgacgctcttccgatctcaaaatggttctggatcagct-3′,
hras_r1(seqidno.16):
5′-gacgtgtgctcttccgatctcctgccagcagctgccctgtgg-3′;
hras_f2(seqidno.17):
5′-acacgacgctcttccgatctgtcattgatggggagacgtgc-3′,
hras_r2(seqidno.18):
5′-gacgtgtgctcttccgatctcaaacacacacaggaagccctc-3′;
扩增tp53的引物对:
tp53_f1(seqidno.19):
5′-acacgacgctcttccgatcttttccttactgcctcttgcttct-3′,
tp53_r1(seqidno.20):
5′-gacgtgtgctcttccgatcttgcgccggtctctcccaggac-3′;
扩增tert的引物对:
tert_f1(seqidno.21):
5′-acacgacgctcttccgatctcagcgctgcctgaaactcgc-3′,
tert_r1(seqidno.22):
5′-gacgtgtgctcttccgatctcaccttccagctccgcctcctc-3′;
第二轮扩增通用引物对:
i5xx(seqidno.23):5′-aatgatacggcgaccaccgagatctacac【i5】
acactctttccctacacgacgctcttccgatct-3′,
i7xx(seqidno.24):5′-caagcagaagacggcatacgagat【i7】
gtgactggagttcagacgtgtgctcttccgatct-3′;
*i5和i7为6个碱基序列,index编号不同这6个碱基序列不同,用于分样。
扩增gapdh的引物对:
gapdh_f1(seqidno.25):
5′-acacgacgctcttccgatcttgggggcgatgctggcgctg-3′,
gapdh_r1(seqidno.26):
5′-gacgtgtgctcttccgatcttcattacttctcatggttcac-3′;
扩增krt7的引物对:
krt7_f1(seqidno.27):
5′-acacgacgctcttccgatctccggaatgagatttcagagat-3′,
krt7_r1(seqidno.28):
5′-gacgtgtgctcttccgatctgcacgagcatccttgagcgcc-3′;
扩增ret-ccdc6与ret-ncoa4的引物对:
ret-ccdc6_f1(seqidno.29):
5′-acacgacgctcttccgatctactgcaggaggagaaccgcg-3′,
ret-ncoa4_f1(seqidno.30):
5′-acacgacgctcttccgatctcccaggactggcttatcca-3′;
ret_r1(seqidno.31):
5′-gacgtgtgctcttccgatctgaaggccgttgccttgacca-3′;
扩增pax8-pparg的引物对:
pax8-pparg_7f1(seqidno.32):5′-
acacgacgctcttccgatcttgcccatttgagcggcagc-3′;
pax8-pparg_8f1(seqidno.33):
5′-acacgacgctcttccgatctgacgggaaggccaccctg-3′;
pax8-pparg_9f1(seqidno.34):
5′-acacgacgctcttccgatctgtcccgcccttcaatgcct-3′;
pax8-pparg_10f1(seqidno.35):
5′-acacgacgctcttccgatctacatccccaccagcggaca-3′;
pax8-pparg_r1(seqidno.36):
5′-gacgtgtgctcttccgatctgagatccacggagctgatcc-3′。
实施例2:试剂盒的制备方法。
(1)高保真pcr酶混合液:该反应液中含有酶缓冲液、mg2+、dntps、高保真dna聚合酶,-20℃保存;
(2)dna特异性引物混合池:该反应液中含有dna扩增特异性引物,包含seqidno.1~22,浓度为10um,-20℃保存;
(3)rna特异性引物混合池:该反应液中含有cdna扩增特异性引物,包含seqidno.25~36,浓度为10um,-20℃保存;
(4)i5xx引物:该反应液中含有带i5xxindex的通用引物,浓度为10um,-20℃保存;
(5)i7xx引物:该反应液中含有带i7xxindex的通用引物,浓度为10um,-20℃保存;
(6)阴性对照:去rna酶的水。
实施例3:检测方法。
仪器:定性pcr仪、涡旋混匀仪、24孔高速离心机、qubit荧光定量仪、安捷伦4200片段分析仪、纯水仪、二代高通量测序仪。
(1)样本dna和rna共提取:使用qiagenallprepdna/rnaffpe核酸提取试剂盒(货号:80234),参考试剂盒说明书进行操作,提取样本中的dna和rna,样本dna作为核酸恒温扩增反应模板备用。
(2)样本rna进行逆转录制备cdna:提取好的rna样本采用takaraprimescripttmrtreagentkitwithgdnaeraser(perfectrealtime)(货号:rr047a),参考试剂盒说明书进行操作,逆转录成cdna,cdna作为核酸恒温扩增反应模板备用。
(3)使用本试剂盒进行文库构建
(3a)dna靶区域扩增
按照下列反应体系和扩增条件进行第一轮pcr扩增,使用0.2mlpcr管/8连管,在超净台里按照如下体系配制反应:
dna多重pcr扩增程序设置:
(3b)dna第一轮pcr产物纯化:
利用ampurexpbeads纯化试剂盒或其他功能等效的试剂盒对pcr产物进行纯化。该纯化可有效的富集目标区域片段,降低非特异性产物的量。
1)将以上20μl扩增产物,涡旋混匀,并瞬时离心,然后加入20μlampurexp磁珠(需事先从冰箱中取出并平衡至室温后使用),涡旋混匀,室温放置5分钟;
2)以上离心管瞬时离心,放置于磁力架上,静置5分钟,待溶液澄清后,小心吸去上清(避免吸起磁珠);
3)然后加入100~200μl80%的乙醇,磁力架反复(不少于两次)在1.5ml离心管不同的两面来回吸附磁珠(即在磁力架上旋转1.5ml离心管,使得磁珠在80%乙醇中来回游移),以充分洗涤磁珠,然后在磁力架上,静置1分钟;
4)小心吸去上清(避免吸起磁珠),再次加入100~200μl80%的无水乙醇,重复步骤“3)”;
5)用移液枪小心吸除上清,尽可能吸取干净(避免吸起磁珠),然后将1.5ml离心管置于通风橱中,静置5分钟,使乙醇挥发完全;
注:乙醇必须去除干净,否则残留的乙醇会严重影响得率和后续实验;
6)向离心管中加入20μl去离子水,瞬时涡旋,充分悬浮磁珠,室温放置5分钟,无需去除磁珠,直接进行第二轮扩增。
(3c)dna第二轮pcr扩增:
对纯化产物进行第二轮pcr扩增,使用测序通用引物i5xx引物与i7xx引物进行,使用0.2mlpcr管/8连管,在超净台里按照如下体系配制反应:
*不同的样本文库请使用不同的index编号引物
dna多重pcr扩增程序设置:
(3d)dna第二轮pcr产物回收
利用ampurexpbeads纯化试剂盒或其他功能等效的试剂盒对pcr产物进行纯化。
1)将以上50μl扩增产物,涡旋混匀,并瞬时离心,然后加入40μlampurexp磁珠(需事先从冰箱中取出并平衡至室温后使用),涡旋混匀,室温放置5分钟;
2)以上离心管瞬时离心,放置于磁力架上,静置5分钟,待溶液澄清后,小心吸去上清(避免吸起磁珠);
3)然后加入100~200μl80%的乙醇,磁力架反复(不少于两次)在1.5ml离心管不同的两面来回吸附磁珠(即在磁力架上旋转1.5ml离心管,使得磁珠在80%乙醇中来回游移),以充分洗涤磁珠,然后在磁力架上,静置1分钟;
4)小心吸去上清(避免吸起磁珠),再次加入100~200μl80%的无水乙醇,重复步骤“3)”;
5)用移液枪小心吸除上清,尽可能吸取干净(避免吸起磁珠),然后将1.5ml离心管置于通风橱中,静置5分钟,使乙醇挥发完全;
注:乙醇必须去除干净,否则残留的乙醇会严重影响得率和后续实验;
6)向离心管中加入20μl去离子水,瞬时涡旋,充分悬浮磁珠,室温放置5分钟;
7)将以上离心管瞬时离心,置于磁力架上,静置5分钟,直至溶液澄清,小心吸取20μl上清,至新的1.5ml离心管中,该纯化产物即为构建好的文库,尽快上机测序。(3e)cdna靶区域扩增
按照下列反应体系和扩增条件进行第一轮pcr扩增。使用0.2mlpcr管/8连管,在超净台里按照如下体系配制反应:
cdna多重pcr扩增程序设置:
(3f)cdna第一轮pcr产物纯化
利用ampurexpbeads纯化试剂盒或其他功能等效的试剂盒对pcr产物进行纯化。该纯化可有效的富集目标区域片段,降低非特异性产物的量。
1)将以上20μl扩增产物,涡旋混匀,并瞬时离心,然后加入20μlampurexp磁珠(需事先从冰箱中取出并平衡至室温后使用),涡旋混匀,室温放置5分钟;
2)以上离心管瞬时离心,放置于磁力架上,静置5分钟,待溶液澄清后,小心吸去上清(避免吸起磁珠);
3)然后加入100~200μl80%的乙醇,磁力架反复(不少于两次)在1.5ml离心管不同的两面来回吸附磁珠(即在磁力架上旋转1.5ml离心管,使得磁珠在80%乙醇中来回游移),以充分洗涤磁珠,然后在磁力架上,静置1分钟;
4)小心吸去上清(避免吸起磁珠),再次加入100~200μl80%的无水乙醇,重复步骤“3)”;
5)用移液枪小心吸除上清,尽可能吸取干净(避免吸起磁珠),然后将1.5ml离心管置于通风橱中,静置5分钟,使乙醇挥发完全;
注:乙醇必须去除干净,否则残留的乙醇会严重影响得率和后续实验;
6)向离心管中加入20μl去离子水,瞬时涡旋,充分悬浮磁珠,室温放置5分钟,无需去除磁珠,直接进行第二轮扩增。
(3g)cdna第二轮pcr扩增
对纯化产物进行第二轮pcr扩增,使用0.2mlpcr管/8连管,在超净台里按照如下体系配制反应:
*不同的文库请使用不同的index编号引物
cdna多重pcr扩增程序设置:
(3h)cdna第二轮pcr产物回收
利用ampurexpbeads纯化试剂盒或其他功能等效的试剂盒对pcr产物进行纯化。
1)将以上50μl扩增产物,涡旋混匀,并瞬时离心,然后加入40μlampurexp磁珠(需事先从冰箱中取出并平衡至室温后使用),涡旋混匀,室温放置5分钟;
2)以上离心管瞬时离心,放置于磁力架上,静置5分钟,待溶液澄清后,小心吸去上清(避免吸起磁珠并尽量洗净上清);
3)向八连管或pcr管中加入20ul去离子水,瞬时涡旋,瞬时离心,室温放置5分钟;
4)以上离心管瞬时离心,放置于磁力架上,静置5分钟,待溶液澄清后,小心将上清(避免吸起磁珠)转移至新的离心管中;
5)然后向以上离心管中加入16μlampurexp磁珠,涡旋混匀,室温放置5分钟;
6)然后加入100~200μl80%的乙醇,磁力架反复(不少于两次)在1.5ml离心管不同的两面来回吸附磁珠(即在磁力架上旋转1.5ml离心管,使得磁珠在80%乙醇中来回游移),以充分洗涤磁珠,然后在磁力架上,静置1分钟;
7)小心吸去上清(避免吸起磁珠),再次加入100~200μl80%的无水乙醇,重复步骤“6)”;
8)用移液枪小心吸除上清,尽可能吸取干净(避免吸起磁珠),然后将1.5ml离心管置于通风橱中,静置5分钟,使乙醇挥发完全;
注:乙醇必须去除干净,否则残留的乙醇会严重影响得率和后续实验;
9)向离心管中加入20μl去离子水,瞬时涡旋,充分悬浮磁珠,室温放置5分钟;
10)将以上离心管瞬时离心,置于磁力架上,静置5分钟,直至溶液澄清,小心吸取20μl上清,至新的1.5ml离心管中,该纯化产物即为构建好的文库,尽快上机测序。
(4)上机测序:将建好的文库,准确定量并稀释到合适浓度,参考illunima官方操作说明书,将每个样本的dna扩增文库和cdna扩增文库各分配40m数据量进行上机测序。
(5)生信分析:利用以下第三方软件:bcl2fastq,ngsqctoolkit,fastqc,fastp,speedseq,genomeanalysistoolkit(gatk),samtools,varscan2,snpeff,bamdst进行数据结果分析。
实施例4:癌组织和癌旁组织一对一回顾性检测。
将本发明所述的试剂盒用于回顾性检测10例手术后甲状腺癌患者,即将手术切除的10例癌组织和10例癌旁组织进行检测,此10例患者的20例样本由杭州市邵逸夫医院头颈科提供,检测方法按照实施例3中的步骤实施,检测结果与邵逸夫医院病理科医生对20例样品的病理诊断结果进行比对,检测结果表1所示:
表1回顾性检测结果对照表
由上表可见,10例甲状腺癌患者的肿瘤组织中都检测出甲状腺癌致病相关基因的变异,与病理诊断结果相吻合,具有100%的阳性符合率;10例甲状腺癌患者的癌旁正常组织则没有检测到变异,与病理诊断的结果相一致,具有100%的阴性符合率。由此回顾性检测实例结果可见:本试剂盒检测结果具有较高的特异性。
实施例5:临床患者甲状腺结节样本检测。
将本发明所述的试剂盒用于20例甲状腺结节样本的检测,此20例样本由杭州市邵逸夫医院头颈科提供,检测方法按照实施例3中的步骤实施,检测结果与病理医生对20例样本的病理诊断结果进行比对,检测结果表2所示:
表2样本检测结果对照表
由上表可见:20例甲状腺病变患者,其中9例给出明确的病理诊断结果为甲状腺癌,这9例样本本试剂盒的检测结果显示均存在甲状腺癌相关基因的变异,与病理诊断结果相一致;其中11例病理诊断无法给出明确的良恶性结果,这11例样本本试剂盒的检测结果显示7例检测出甲状腺癌相关基因的变异,4例没有检出。由此临床样本检测实例结果可见:本试剂盒的检测结果为进一步诊断甲状腺病变类型提供非常重要的参考依据。
序列表
<110>杭州迪安医学检验中心有限公司
<120>一种基于高通量测序技术用于检测甲状腺癌致病相关基因变异的引物及其应用
<160>36
<170>siposequencelisting1.0
<210>1
<211>41
<212>dna
<213>人工序列(artificialsequence)
<400>1
<210>2
<211>42
<212>dna
<213>人工序列(artificialsequence)
<400>2
<210>3
<211>42
<212>dna
<213>人工序列(artificialsequence)
<400>3
<210>4
<211>41
<212>dna
<213>人工序列(artificialsequence)
<400>4
<210>5
<211>43
<212>dna
<213>人工序列(artificialsequence)
<400>5
<210>6
<211>42
<212>dna
<213>人工序列(artificialsequence)
<400>6
<210>7
<211>41
<212>dna
<213>人工序列(artificialsequence)
<400>7
<210>8
<211>42
<212>dna
<213>人工序列(artificialsequence)
<400>8
<210>9
<211>41
<212>dna
<213>人工序列(artificialsequence)
<400>9
<210>10
<211>42
<212>dna
<213>人工序列(artificialsequence)
<400>10
<210>11
<211>41
<212>dna
<213>人工序列(artificialsequence)
<400>11
<210>12
<211>41
<212>dna
<213>人工序列(artificialsequence)
<400>12
<210>13
<211>41
<212>dna
<213>人工序列(artificialsequence)
<400>13
<210>14
<211>42
<212>dna
<213>人工序列(artificialsequence)
<400>14
<210>15
<211>41
<212>dna
<213>人工序列(artificialsequence)
<400>15
<210>16
<211>42
<212>dna
<213>人工序列(artificialsequence)
<400>16
<210>17
<211>41
<212>dna
<213>人工序列(artificialsequence)
<400>17
<210>18
<211>42
<212>dna
<213>人工序列(artificialsequence)
<400>18
<210>19
<211>43
<212>dna
<213>人工序列(artificialsequence)
<400>19
<210>20
<211>41
<212>dna
<213>人工序列(artificialsequence)
<400>20
<210>21
<211>40
<212>dna
<213>人工序列(artificialsequence)
<400>21
<210>22
<211>42
<212>dna
<213>人工序列(artificialsequence)
<400>22
<210>23
<211>68
<212>dna
<213>人工序列(artificialsequence)
<400>23
<210>24
<211>64
<212>dna
<213>人工序列(artificialsequence)
<400>24
<210>25
<211>40
<212>dna
<213>人工序列(artificialsequence)
<400>25
<210>26
<211>41
<212>dna
<213>人工序列(artificialsequence)
<400>26
<210>27
<211>41
<212>dna
<213>人工序列(artificialsequence)
<400>27
<210>28
<211>41
<212>dna
<213>人工序列(artificialsequence)
<400>28
<210>29
<211>40
<212>dna
<213>人工序列(artificialsequence)
<400>29
<210>30
<211>39
<212>dna
<213>人工序列(artificialsequence)
<400>30
<210>31
<211>40
<212>dna
<213>人工序列(artificialsequence)
<400>31
<210>32
<211>39
<212>dna
<213>人工序列(artificialsequence)
<400>32
<210>33
<211>38
<212>dna
<213>人工序列(artificialsequence)
<400>33
<210>34
<211>39
<212>dna
<213>人工序列(artificialsequence)
<400>34
<210>35
<211>39
<212>dna
<213>人工序列(artificialsequence)
<400>35
<210>36
<211>40
<212>dna
<213>人工序列(artificialsequence)
<400>36
- 还没有人留言评论。精彩留言会获得点赞!