N-乙烯基-1,2,3-三氮唑类化合物的制备方法与流程
本发明属于有机合成化学技术领域,尤其涉及n-乙烯基-1,2,3-三氮唑类化合物的制备方法。
背景技术:
1,2,3-三氮唑及其衍生物,有着十分广泛的应用,包括有机合成(angew.chem.int.ed,,2014,53,3868),材料科学(chem.rev,2016,116,5689),生物化学(chem.rev.,2016,116,3086)和药物合成(j.am.chem.soc.,2017,139,3756)。作为它们的一部分,n-乙烯基取代的1,2,3-三氮唑是工业官能化聚合物的重要前体。许多化学工作者利用n-乙烯基-1,2,3-三氮唑易聚合性质大规模的生产富电子聚合物(polym.chem.,2012,3,1680)。目前,制备n-乙烯基-1,2,3-三氮唑的有两种通用的方法,一是在合适的条件下用亲电试剂对nh/磺酰基-三氮唑进行后修饰(bioorg.med.chem.lett.,2009,19,3899;org.biomol.chem.,2017,15,2721);二是通过碱介导的体系,具有活性亚甲基,炔,1,3-二羰基片段或α-酮基膦的乙烯基叠氮化合物环加成(greenchem.2015,17,781;newj.chem.2016,40,6559;eur.j.org.chem.2017,459)。这些合成方法广泛应用受到有限的底物范围,冗余的前期预合成步骤,反应试剂易爆炸和有毒性的束缚。因此,获得乙烯基三氮唑的有效合成方法是非常需要和迫切的。
随着sharpless(angew.chem.int.ed.,2002,41,2596)和meldal(j.org.chem.,2002,67,3057)等人提出铜催化点击化学的概念,以乙烯基叠氮化作为反应底物,铜催化的叠氮-炔烃环加成(cuaac)和随后钌(ii)催化叠氮-炔烃环加成(ruaac)已经成为高效的和区域选择性构建n-乙烯基取代的1,2,3-三氮唑的策略。然而,由于叠氮化合物本身具有潜在的爆炸性,乙烯基叠氮化合物的取代基局限于芳基或长碳烷基。在设计用于该策略的有机叠氮化合物时,应该考虑有机叠氮化合物中的所有氮原子。为了获得具有爆炸性的2-叠氮丙烯或类似物,低温和强碱是必不可少的。同时,由于三氮唑的药物性质与取代类型密切相关,所提到的这些局限性损害了乙烯基-1,2,3-三氮唑类化合物在药物化学中的重要作用。
技术实现要素:
针对上述现有技术中存在的问题,本发明旨在提供n-乙烯基-1,2,3-三氮唑类化合物的制备方法。本发明设计了具有爆炸性的2-叠氮丙烯或类似物,可以通过这种保护-脱保护策略原位产生,并且在没有进一步分离的情况下快速地与适当的底物进行反应。在此基础上,本发明通过原位生成2-叠氮丙烯及其衍生物的方法获得n-乙烯基-1,2,3-三氮唑,该方法操作极其简单、底物和催化剂简便易得、底物普适性优良,良好的官能团耐受性和对环境大气的不敏感性,具有均匀的良好至极好的产率。
本发明的目的之一是提供n-乙烯基-1,2,3-三氮唑类化合物的制备方法。
本发明的目的之二是提供上述n-乙烯基-1,2,3-三氮唑类化合物的制备方法的应用。
为实现上述发明目的,具体的,本发明公开了下述技术方案:
首先,本发明公开n-乙烯基-1,2,3-三氮唑类化合物的制备方法,其反应原理如式(1)所示;包括将端基炔类化合物1、化合物2、亲核试剂nu以及pd/cu催化剂加入含有三乙胺的溶剂中,在氩气保护下加热反应,得到n-乙烯基-1,2,3-三氮唑类化合物3。
所述反应式(1)中,r1可为富电子或缺电子芳基、杂芳基、烷基;nr2可为吗啉、哌啶、二乙胺、四氢吡咯、苯胺、甲基苯胺、邻苯二甲酰亚胺、吲哚、二异丙胺、l-脯氨酸、苄胺、n甲基甲酰胺、二甲胺、二苯胺、n乙酰苯胺、1-萘胺、炔丙胺、n甲基炔丙基胺、正丁胺、环己胺等。
具体的,所述n-乙烯基-1,2,3-三氮唑类化合物的制备方法,合成路线如式(2)所示:
具体的,n-乙烯基-1,2,3-三氮唑类化合物的制备包括如下步骤:
(1)将端基炔类化合物1、化合物2以及亲核试剂nu加入含有三乙胺的溶剂中,随后向反应体系中加入pd/cu催化剂,在氩气保护下加热反应,底物消失后反应结束;
(2)将步骤(1)得到的反应液减压蒸馏除去溶剂,将得到的液体进行洗脱,得到n-乙烯基-1,2,3-三氮唑类化合物。
步骤(1)中,所述端基炔类化合物1与化合物2的摩尔比为(0.8-2):1;优选为1.5:1。
步骤(1)中,所述催化剂为pd(pph3)4或pd(oac)2中的任意一种和cui、cucl2、cu(oac)2中的任意一种的混合物。
步骤(1)中,所述亲核试剂nu包括nabh4。
步骤(1)中,所述催化剂中,pd(pph3)4的添加量为化合物2的添加量的1-5%;cui、cucl2或cu(oac)2的添加量为化合物2的添加量的2-8%;优选为pd(pph3)4为2%,cui、cucl2或cu(oac)2为4%。
步骤(1)中,所述加热反应的温度为35-80℃,优选为60℃。
步骤(1)中,所述溶剂可以选择水、1,2-二氯乙烷、甲醇、乙醇、甲苯、硝基甲烷、n,n-二甲基甲酰胺(dmf)、乙二醇、二甲基亚砜(dmso),乙腈中的任意一种或与三乙胺的混合物;优选的,所述混合物中前、后两者的体积比为1:0.2-1.5。
更优选地,所述溶剂由体积比1:1的乙腈、三乙胺的混合液构成。
步骤(1)中,所述洗脱用硅胶柱层析的方法,其中,洗脱液为v石油醚:v乙酸乙酯=3-6:1,优选为5:1。
最后,本发明公开上述n-乙烯基-1,2,3-三氮唑类化合物的制备方法在生物、医药等领域中的应用。
与现有技术相比,本发明取得的有益效果是:
(1)本发明操作简单,原料和试剂易得,条件温和,反应体系绿色环保,产物易分离纯化,可以通过一锅法高效、高收率地合成各种高度官能化的乙烯基-1,2,3-三氮唑类化合物。
(2)本发明设计了具有爆炸性的2-叠氮丙烯或类似物,通过这种保护-脱保护策略原位产生,并且在没有进一步分离的情况下快速地与适当的底物进行反应。在此基础上,本发明通过原位生成2-叠氮丙烯及其衍生物的方法获得n-乙烯基-1,2,3-三氮唑,该方法操作极其简单、底物和催化剂简便易得、底物普适性优良,良好的官能团耐受性和对环境大气的不敏感性,具有均匀的良好至极好的产率。
附图说明
构成本申请的一部分的说明书附图用来提供对本申请的进一步理解,本申请的示意性实施例及其说明用于解释本申请,并不构成对本申请的不当限定。
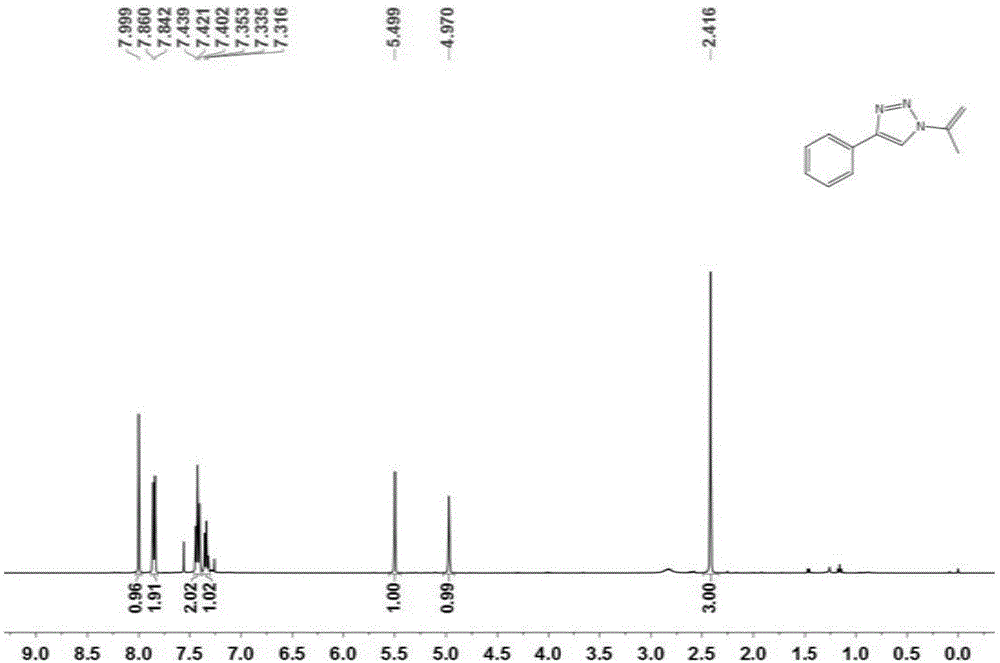
图1为实施例1制备的化合物3a的1h-nmr的核磁共振谱。
图2为实施例1制备的化合物3a的13c-nmr的核磁共振谱。
图3为实施例10制备的化合物3b的1h-nmr的核磁共振谱。
图4为实施例10制备的化合物3b的13c-nmr的核磁共振谱。
图5为实施例11制备的化合物3c的1h-nmr的核磁共振谱。
图6为实施例11制备的化合物3c的13c-nmr的核磁共振谱。
图7为实施例12制备的化合物3d的1h-nmr的核磁共振谱。
图8为实施例12制备的化合物3d的13c-nmr的核磁共振谱。
图9为实施例13制备的化合物3e的1h-nmr的核磁共振谱。
图10为实施例13制备的化合物3e的13c-nmr的核磁共振谱。
图11为实施例14制备的化合物3f的1h-nmr的核磁共振谱。
图12为实施例14制备的化合物3f的13c-nmr的核磁共振谱。
图13为实施例15制备的化合物3g的1h-nmr的核磁共振谱。
图14为实施例15制备的化合物3g的13c-nmr的核磁共振谱。
图15为实施例16制备的化合物3h的1h-nmr的核磁共振谱。
图16为实施例16制备的化合物3h的13c-nmr的核磁共振谱。
图17为实施例17制备的化合物3i的1h-nmr的核磁共振谱。
图18为实施例17制备的化合物3i的13c-nmr的核磁共振谱。
图19为实施例18制备的化合物3j的1h-nmr的核磁共振谱。
图20为实施例18制备的化合物3j的13c-nmr的核磁共振谱。
具体实施方式
应该指出,以下详细说明都是例示性的,旨在对本申请提供进一步的说明。除非另有指明,本文使用的所有技术和科学术语具有与本申请所属技术领域的普通技术人员通常理解的相同含义。
需要注意的是,这里所使用的术语仅是为了描述具体实施方式,而非意图限制根据本申请的示例性实施方式。如在这里所使用的,除非上下文另外明确指出,否则单数形式也意图包括复数形式,此外,还应当理解的是,当在本说明书中使用术语“包含”和/或“包括”时,其指明存在特征、步骤、操作、器件、组件和/或它们的组合。
正如背景技术所介绍的,由于叠氮化合物本身具有潜在的爆炸性,乙烯基叠氮化合物的取代基局限于芳基或长碳烷基。在设计用于该策略的有机叠氮化合物时,应该考虑有机叠氮化合物中的所有氮原子。为了获得具有爆炸性的2-叠氮丙烯或类似物,低温和强碱是必不可少的。同时,由于三氮唑的药物性质与取代类型密切相关,所提到的这些局限性损害了乙烯基-1,2,3-三氮唑类化合物在药物化学中的重要作用。为此,本发明提出一n-乙烯基-1,2,3-三氮唑类化合物的制备方法,下面结合附图和具体实施方式对本发明进一步说明。
实施例1
一种n-乙烯基-1,2,3-三氮唑类化合物的制备方法,反应过程如式(3):
反应具体包括如下步骤:
(1)将化合物1a即苯乙炔(34ul,0.3mmol)、化合物2(82mg,0.2mmol)、nabh4(3.8mg,0.4mmol)、加入到1ml乙腈和1ml三乙胺中,随后向反应体系中加入催化剂pd(pph3)4(4.7mg,0.004mmol)和cui(1.5mg,0.008mmol),氩气保护,60℃下加热搅拌4h,tlc检测底物消失,反应结束;将反应液减压蒸馏除去有机溶剂,得到粘稠的液体;
(2)将步骤(1)得到的粘稠液体经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=5:1)得到化合物3a,其产率高达91%。
本实施例制备的化合物3a的1h-nmr和13cnmr的核磁共振谱分别如图1和2所示,结果为:
1hnmr(400mhz,cdcl3)δ8.00(s,1h),7.88–7.82(m,2h),7.45–7.39(t,j=8.3hz,2h),7.36–7.30(m,1h),5.50(s,1h),4.97(s,1h),2.42(s,3h).
13cnmr(101mhz,cdcl3)δ147.72,139.26,130.29,128.87,128.34,125.84,116.77,103.75,19.47.hrms(esi)m/zcalculatedforc11h11n3[m+h]+:186.1031,found186.1121.
试验例1
一种n-乙烯基-1,2,3-三氮唑类化合物的制备方法,反应过程如实施例1中式(3);区别在于:
(1)将化合物1a即苯乙炔(34ul,0.3mmol)、化合物2(82mg,0.2mmol)、nabh4(3.8mg,0.4mmol)、加入到1ml乙腈和1ml三乙胺中,随后向体系中加入催化剂pd(pph3)4(4.7mg,0.004mmol)和cui(1.5mg,0.008mmol),氩气保护,32℃下加热搅拌4h,直到tlc检测底物消失,反应结束;将反应液减压蒸馏除去有机溶剂,得到粘稠的液体;
(2)将步骤(1)得到的粘稠液体经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=5:1),检测到化合物3a的产率为0%,即在本试验例的反应条件下未能合成n-乙烯基-1,2,3-三氮唑类化合物。
实施例2
一种n-乙烯基-1,2,3-三氮唑类化合物的制备方法,反应过程如实施例1中式(3);区别在于:
(1)将化合物1a即苯乙炔(0.2mmol)、化合物2(82mg,0.2mmol)、nabh4(3.8mg,0.4mmol)、加入到1ml水和0.8ml三乙胺中,随后向体系中加入催化剂pd(pph3)4(0.008mmol)和cui(0.004mmol),氩气保护,80℃下加热搅拌4h,直到tlc检测底物消失,反应结束;将反应液减压蒸馏除去有机溶剂,得到粘稠的液体;
(2)将步骤(1)得到的粘稠液体经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=5:1)得到化合物3a,其产率为65%。
实施例3
一种n-乙烯基-1,2,3-三氮唑类化合物的制备方法,反应过程如实施例1中式(3);区别在于:
(1)将化合物1a即苯乙炔(34ul,0.3mmol)、化合物2(82mg,0.2mmol)、nabh4(3.8mg,0.4mmol)、加入到1ml甲醇和1ml三乙胺中,随后向体系中加入催化剂pd(pph3)4(0.004mmol)和cucl2(1.4mg,0.008mmol),氩气保护,60℃下加热搅拌4h,直到tlc检测底物消失,反应结束;将反应液减压蒸馏除去有机溶剂,得到粘稠的液体;
(2)将步骤(1)得到的粘稠液体经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=5:1)得到化合物3a,其产率高达88%。
试验例2
一种n-乙烯基-1,2,3-三氮唑类化合物的制备方法,反应过程如实施例1中式(3);区别在于:
(1)将化合物1a即苯乙炔(34ul,0.3mmol)、化合物2(82mg,0.2mmol)、nabh4(3.8mg,0.4mmol)、加入到1ml甲醇和1ml三乙胺,随后向体系中加入催化剂pd(pph3)4(4.7mg,0.004mmol)和cu2o(1.2mg,0.008mmol),氩气保护,60℃下加热搅拌4h,直到tlc检测底物消失,反应结束;将反应液减压蒸馏除去有机溶剂,得到粘稠的液体;
(2)将步骤(1)得到的粘稠液体经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=5:1),检测到化合物3a的产率为0%,即在本试验例的反应条件下未能合成n-乙烯基-1,2,3-三氮唑类化合物。
实施例4
一种n-乙烯基-1,2,3-三氮唑类化合物的制备方法,反应过程如实施例1中式(3);区别在于:
(1)将化合物1a即苯乙炔(0.16mmol)、化合物2(0.2mmol)、nabh4(0.4mmol)、加入到1ml甲苯和1.5ml三乙胺中,随后向体系中加入催化剂pd(pph3)4(0.002mmol)和cu(oac)2(0.008mmol),氩气保护,60℃下加热搅拌4h,直到tlc检测底物消失,反应结束;将反应液减压蒸馏除去有机溶剂,得到粘稠的液体;
(2)将步骤(1)得到的粘稠液体经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=5:1)得到化合物3a,其产率高达84%。
实施例5
一种n-乙烯基-1,2,3-三氮唑类化合物的制备方法,反应过程如实施例1中式(3);区别在于:
(1)将化合物1a即苯乙炔(0.18mmol)、化合物2(0.2mmol)、nabh4(0.4mmol)、加入到1ml1,2-二氯乙烷和0.5ml三乙胺中,随后向体系中加入催化剂pd(pph3)2cl2(0.009mmol)和cui(0.016mmol),氩气保护,60℃下加热搅拌4h,直到tlc检测底物消失,反应结束;将反应液减压蒸馏除去有机溶剂,得到粘稠的液体;
(2)将步骤(1)得到的粘稠液体经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=5:1)得到化合物3a的产率为63%。
实施例6
一种n-乙烯基-1,2,3-三氮唑类化合物的制备方法,反应过程如实施例1中式(3);区别在于:
(1)将化合物1a即苯乙炔(0.25mmol)、化合物2(0.2mmol)、nabh4(0.4mmol)、加入到1ml乙二醇和0.2ml三乙胺中,随后向体系中加入催化剂pd(oac)2(0.01mmol)和cui(0.012mmol),氩气保护,65℃下加热搅拌4h,直到tlc检测底物消失,反应结束;将反应液减压蒸馏除去有机溶剂,得到粘稠的液体;
(2)将步骤(1)得到的粘稠液体经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=3:1)得到化合物3a的产率为23%。
实施例7
一种n-乙烯基-1,2,3-三氮唑类化合物的制备方法,反应过程如实施例1中式(3);区别在于:
(1)将化合物1a即苯乙炔(0.3mmol)、化合物2(0.2mmol)、nabh4(0.4mmol)、加入到1ml硝基甲烷和1ml三乙胺中,随后向体系中加入催化剂pd(pph3)4(0.004mmol)和cui(0.006mmol),氩气保护,35℃下加热搅拌5h,直到tlc检测到底物消失,反应结束;将反应液减压蒸馏除去有机溶剂,得到粘稠的液体;
(2)将步骤(1)得到的粘稠液体经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=5:1)得到化合物3a的最高产率55%。
实施例8
一种n-乙烯基-1,2,3-三氮唑类化合物的制备方法,反应过程如实施例1中式(3);区别在于:
(1)将化合物1a即苯乙炔(0.3mmol)、化合物2(0.15mmol)、nabh4(0.4mmol)、加入到1ml二甲基亚砜和1.2mlml三乙胺中,随后向体系中加入催化剂pd(pph3)4(0.004mmol)和cui(0.008mmol),氩气保护,40℃下加热搅拌5h,直到tlc检测底物消失,反应结束。将反应液减压蒸馏除去有机溶剂,得到粘稠的液体;
(2)将步骤(1)得到的粘稠液体经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=6:1)得到化合物3a的最高产率66%。
实施例9
一种n-乙烯基-1,2,3-三氮唑类化合物的制备方法,反应过程如实施例1中式(3);区别在于:
(1)将化合物1a即苯乙炔(0.3mmol)、化合物2(0.2mmol)、nabh4(0.4mmol)、加入到1ml乙二醇和0.6ml三乙胺中,随后向体系中加入催化剂pd(pph3)4(0.004mmol)和cui(0.008mmol),氩气保护,50℃下加热搅拌4h,直到tlc检测底物消失,反应结束;将反应液减压蒸馏除去有机溶剂,得到粘稠的液体;
(2)将步骤(1)得到的粘稠液体经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=5:1)得到化合物3a的最高产率47%。
实施例10
一种n-乙烯基-1,2,3-三氮唑类化合物的制备方法,反应过程如下列式(4);区别在于:(1)用4-氯苯乙炔(即化合物1b)代替实施例1中的化合物1a,(2)所述溶剂为体积比为1:1的n,n-二甲基甲酰胺和三乙胺组成的混合物,其他条件同实施例1,得到化合物3b产率为95%。
本实施例制备的化合物3b的1h-nmr和13cnmr的核磁共振谱分别如图3和4所示,结果为:
1hnmr(400mhz,cdcl3)δ7.99(s,1h),7.80(d,j=8.5hz,2h),7.41(d,j=8.6hz,2h),5.51(s,1h),5.00(s,1h),2.44(s,3h).
13cnmr(101mhz,cdcl3)δ146.69,139.22,134.13,129.10,128.80,127.07,116.81,103.98,19.47.hrms(esi)m/zcalculatedforc11h10cln3[m+h]+:220.0641,found220.0612.
实施例11
一种n-乙烯基-1,2,3-三氮唑类化合物的制备方法,反应过程如下列式(5);区别在于:用3-甲基苯乙炔(即化合物1c)代替实施例1中的化合物1a,其他条件同实施例1,得到化合物3c产率为96%。
本实施例制备的化合物3c的1h-nmr和13cnmr的核磁共振谱分别如图5和6所示,结果为:
1hnmr(400mhz,cdcl3)δ7.99(s,1h),7.71(s,1h),7.66–7.61(m,1h),7.32(t,j=7.6hz,1h),7.16(d,j=7.6hz,1h),5.50(s,1h),4.98(s,1h),2.43(s,3h),2.41(s,3h).
13cnmr(101mhz,cdcl3)δ147.83,139.28,138.56,130.15,129.12,128.76,126.52,122.94,116.72,103.67,21.45,19.47.hrms(esi)m/zcalculatedforc12h13n3[m+h]+:200.1187,found200.1212.
实施例12
一种n-乙烯基-1,2,3-三氮唑类化合物的制备方法,反应过程如下列式(6);区别在于:用3-乙炔基噻吩(即化合物1d)代替实施例1中的化合物1a,其他条件同实施例1,得到化合物3d产率为94%。
本实施例制备的化合物3d的1h-nmr和13cnmr的核磁共振谱分别如图7和8所示,结果为:
1hnmr(400mhz,cdcl3)δ7.90(s,1h),7.73–7.72(m,1h),7.48–7.47(m,1h),7.41–7.39(m,1h),5.49(s,1h),4.98(s,1h),2.43(s,3h).
13cnmr(101mhz,cdcl3)δ143.91,139.24,131.49,126.41,125.81,121.47,116.53,103.70,19.47.hrms(esi)m/zcalculatedforc9h9n3s[m+h]+:192.0595,found192.0538.
实施例13
一种n-乙烯基-1,2,3-三氮唑类化合物的制备方法,反应过程如下列式(7);区别在于:用1己炔(即化合物1e)代替实施例1中的化合物1a,其他条件同实施例1,得到化合物3e产率为81%。
本实施例制备的化合物3e的1h-nmr和13cnmr的核磁共振谱分别如图9和10所示,结果为:
1hnmr(400mhz,cdcl3)δ7.52(s,1h),5.38(s,1h),4.90(s,1h),2.81–2.67(m,2h),2.38(s,3h),1.67(t,j=7.7hz,2h),1.45–1.34(m,2h),0.94(t,j=7.3hz,3h).
13cnmr(101mhz,cdcl3)δ148.51,139.39,117.88,102.91,31.49,25.28,22.31,19.43,13.83.hrms(esi)m/zcalculatedforc9h15n3[m+h]+:166.1344,found166.1364.
实施例14
一种n-乙烯基-1,2,3-三氮唑类化合物的制备方法,反应过程如下列式(8);区别在于:用化合物1f代替实施例1中的化合物1a,其他条件同实施例1,得到化合物3f产率为97%。
本实施例制备的化合物3f的1h-nmr和13cnmr的核磁共振谱分别如图11和12所示,结果为:
1hnmr(400mhz,cdcl3)δ7.53(s,1h),7.27–7.21(m,2h),6.80(d,j=7.9hz,2h),6.77–6.72(m,1h),5.35(s,1h),4.89(s,1h),4.66(s,2h),3.03(s,3h),2.34(s,3h).
13cnmr(101mhz,cdcl3)δ149.07,145.64,139.23,129.29,118.77,117.23,112.92,103.64,48.72,38.60,19.40.hrms(esi)m/zcalculatedforc13h16n4[m+h]+:229.1453,found229.1465.
实施例15
一种n-乙烯基-1,2,3-三氮唑类化合物的制备方法,反应过程如下列式(9);区别在于:用吗啉代替实施例1中的硼氢化钠,其他条件同实施例1,得到化合物3g产率为93%。
本实施例制备的化合物3g的1h-nmr和13cnmr的核磁共振谱分别如图13和14所示,结果为:
1hnmr(400mhz,cdcl3)δ8.28(s,1h),7.90–7.83(m,2h),7.48–7.42(m,2h),7.36(t,j=7.4hz,1h),5.91(s,1h),5.23(s,1h),3.71(s,4h),3.53(s,2h),2.57(s,4h).
13cnmr(101mhz,cdcl3)δ147.47,138.27,130.37,128.89,128.30,125.81,118.17,108.36,66.94,60.65,53.27.hrms(esi)m/zcalculatedforc15h18n4o[m+h]+:271.1559,found271.1604.
实施例16
一种n-乙烯基-1,2,3-三氮唑类化合物的制备方法,反应过程如下列式(10);区别在于:用哌啶代替实施例1中的硼氢化钠,其他条件同实施例1,得到化合物3h产率为95%。
本实施例制备的化合物3h的1h-nmr和13cnmr的核磁共振谱分别如图15和16所示,结果为:
1hnmr(400mhz,cdcl3)δ8.39(s,1h),7.86(d,j=7.3hz,2h),7.44(t,j=7.6hz,2h),7.35(d,j=7.5hz,1h),5.91(s,1h),5.19(s,1h),3.45(s,2h),2.48(s,4h),1.63–1.52(m,4h),1.45(s,2h).
13cnmr(101mhz,cdcl3)δ147.28,138.87,134.18,130.57,128.85,128.16,125.79,118.57,108.06,61.15,54.22,26.00,24.21.hrms(esi)m/zcalculatedforc16h20n4[m+h]+:269.1766,found269.1839.
实施例17
一种n-乙烯基-1,2,3-三氮唑类化合物的制备方法,反应过程如下列式(11);区别在于:用二乙胺代替实施例1中的硼氢化钠,其他条件同实施例1,得到化合物3i产率为93%。
本实施例制备的化合物3i的1h-nmr和13cnmr的核磁共振谱分别如图17和18所示,结果为:
1hnmr(400mhz,cdcl3)δ8.36(s,1h),7.89–7.82(m,2h),7.47–7.40(m,2h),7.38–7.31(m,1h),5.87(s,1h),5.24(s,1h),3.57(s,2h),2.62(q,j=7.1hz,4h),1.05(t,j=7.1hz,6h).
13cnmr(101mhz,cdcl3)δ147.23,140.32,130.59,128.84,128.14,125.77,118.55,107.68,55.68,46.62,11.64.hrms(esi)m/zcalculatedforc15h20n4[m+h]+:257.1766,found257.1778.
实施例18
一种n-乙烯基-1,2,3-三氮唑类化合物的制备方法,反应过程如下列式(12);区别在于:用四氢吡咯代替实施例1中的硼氢化钠,其他条件同实施例1,得到化合物3j产率为92%。
本实施例制备的化合物3j的1h-nmr和13cnmr的核磁共振谱分别如图19和20所示,结果为:
1hnmr(400mhz,cdcl3)δ8.31(s,1h),7.91–7.84(m,2h),7.48–7.39(m,2h),7.39–7.30(m,1h),5.85(s,1h),5.22(s,1h),3.64(s,2h),2.61(s,4h),1.89–1.73(m,4h).
13cnmr(101mhz,cdcl3)δ147.36,140.22,130.55,128.82,128.17,125.82,118.29,106.93,58.02,53.93,23.66.hrms(esi)m/zcalculatedforc15h18n4[m+h]+:255.1609,found255.1656.
以上所述仅为本申请的优选实施例,并不用于限制本申请,对于本领域的技术人员来说,本申请可以有各种更改和变化。凡在本申请的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本申请的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!