VA@PLGA-CS-HA/OHPMC-HA抗菌可注射水凝胶的制备方法与流程
本发明属于生物医用材料技术领域,具体涉及一种va@plga-cs-ha/ohpmc-ha抗菌可注射水凝胶的制备方法。
背景技术:
水凝胶是一种在水中溶胀、并保持大量水分而又不溶解的聚合物。水凝胶聚合物具有大量吸收水分的特性,大量吸收的水分充斥于聚合物网络中,较大程度地伸展于被交联的大分子链,使整个材料具备了一种流体的性质,这与充盈有大量水性液体的机体组织极其相似,柔软、润湿的表面以及与组织的亲和大大减少了刺激性。可注射水凝胶是指具有一定流动性的、能够通过注射的方法应用的一类水凝胶。可注射水凝胶具有温敏性,当包埋药物注射进入人体后,在体温条件下转变成生物可降解的半固体状凝胶。另外,利用可注射水凝胶还可以填充体腔的缺陷或凹陷部位。近年来,一些新型水凝胶基于其优良的药代动力学以及易于调控的机械性能、易于注射、药物包埋率高和药物定点释放等优点,在药物转运应用中得到广泛关注和深入研究。
微球是一种将药物分子包裹于高分子聚合物内,以实现药物长时间平稳释放为目的药物传递系统,因其缓控释的特点得到了广泛关注。目前,临床应用最广泛有效的抗生素递药系统材料是聚甲基丙烯酸甲酯(pmma),常用的负载抗生素有庆大霉素、妥布霉素和万古霉素等。目前报道的可降解的、已用于局部抗生素缓释的无机和有机载体材料有羟基磷灰石(hap)、磷酸钙骨水泥(cpc)、聚乳酸(pla),聚乳酸聚乙醇酸共聚物(plga),聚己内酯(pcl)、壳聚糖、胶原蛋白、海藻酸盐等。壳聚糖(chitosan,cs)已被证明具有许多优良特性,如:生物粘附性、无毒性、良好的组织相容性、生物降解性、来源广泛性等。透明质酸钠(hyaluronicacidsodiumsalt,has)是皮肤和其它组织中广泛存在的天然生物分子,是构成人体细胞间质、关节滑液等结缔组织的主要成分,在关节腔中几乎以纯态形式存在。它能促进细胞的增殖、分化,清除氧自由基,促进伤口愈合、调节渗透压、润滑、促进细胞修复及改善关节的重要作用。羟丙基甲基纤维素(hpmc)是一种重要的纤维素衍生物,属非离子型、水溶性聚合物,无嗅,无味,无毒,广泛用于口服骨架控释和缓释制剂,作为释放阻滞材料调节药物的释放。
本发明综合hpmc、plga、透明质酸、壳聚糖、万古霉素等材料的优点,制备出了一种抗菌可注射水凝胶va@plga-cs-ha/ohpmc-ha。
技术实现要素:
针对背景技术,本发明的目的在于提供一种va@plga-cs-ha/ohpmc-ha抗菌可注射水凝胶的制备方法。
为了实现本发明的上述目的,本发明采用的技术方案如下:
一种va@plga-cs-ha/ohpmc-ha抗菌可注射水凝胶的制备方法,所述方法包括如下步骤:
(1)将氧化羟丙基羧甲基纤维素(ohpmc)溶解于蒸馏水中,配制质量百分比浓度为1~3wt%的ohpmc溶液;将胺化透明质酸(ha-ada)溶解于蒸馏水中,配制质量百分比浓度为1~3wt%的ha-ada溶液;然后将ohpmc溶液和ha-ada溶液等体积混合均匀,得到ohpmc-ha均匀混合液;
(2)按比例将va@plga-cs-ha微球加入到步骤(1)制得的ohpmc-ha均匀混合液中,混匀成胶,制得本发明所述的va@plga-cs-ha/ohpmc-ha抗菌可注射水凝胶。
进一步地,上述技术方案,步骤(1)所述的氧化羟丙基羧甲基纤维素是将羟丙基羧甲基纤维(hpmc)经高碘酸钠氧化改性后制得,所述氧化改性方法具体如下:
配制质量百分比浓度为8~12%的羟丙基羧甲基纤维水溶液,然后按体积比v羟丙基羧甲基纤维溶液:v高碘酸钠溶液=5:1的比例加入高碘酸钠溶液,避光反应24h,再向反应体系中加入与高碘酸钠溶液等体积的乙二醇,继续搅拌30~45min,蒸馏水透析2~3d、冷冻干燥后制得。
优选地,上述技术方案,所述羟丙基羧甲基纤维水溶液的质量百分比浓度为10%,所述高碘酸钠溶液的质量浓度为100mg/ml。
进一步地,上述技术方案,步骤(1)所述的胺化透明质酸是利用偶氮甲酰胺(ada)将透明质酸胺化改性后制得,所述胺化改性方法具体如下:
配制质量百分比浓度为1~3%的透明质酸溶液,然后按摩尔比1:1的比例加入己二酸二酰肼,完全溶解后调节反应体系ph至4.7~4.8,再加入1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐,混合均匀后继续调节反应体系ph为4.7~4.8,最后室温反应12h,蒸馏水透析2~3d、冷冻干燥后制得。
优选地,上述技术方案,所述透明质酸与1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐的摩尔比为1:2。
进一步地,上述技术方案,步骤(2)所述的va@plga-cs-ha微球与ohpmc-ha水凝胶的用量比为0.1~0.3质量份:1体积份,其中:质量份和体积份之间是以g:ml作为基准。
进一步地,上述技术方案,步骤(1)所述的ohpmc溶液质量百分比浓度优选为2wt%;所述ha-ada溶液质量百分比浓度优选为2wt%。
进一步地,上述技术方案,步骤(2)所述成胶时间优选为3~7min。
进一步地,上述技术方案,步骤(2)所述的va@plga-cs-ha微球采用下述方法制得,具体步骤如下:
(a)将聚乳酸聚乙醇酸共聚物(plga)溶解于二氯甲烷中,配制质量百分比浓度为5~15wt%的plga溶液;将壳聚糖(cs)溶解于冰乙酸中,配制质量百分比浓度为0.5~1.5wt%的壳聚糖溶液,备用;将透明质酸钠(has)溶解于蒸馏水中,配制质量百分比浓度为1~3wt%的透明质酸钠溶液,备用;将万古霉素盐酸盐(va)溶解于蒸馏水中,配制质量百分比浓度为25~100mg/ml的万古霉素溶液;将聚乙烯醇(pva)溶解于蒸馏水中,配制质量百分比浓度为0.3~3wt%的壳聚糖溶液,备用;
(b)按比例将步骤(a)所述plga溶液与va溶液混合均匀后超声分散,形成w1/o乳液,其中:所述plga溶液与va溶液的体积比为2~10:1;
(c)按比例将步骤(a)所述壳聚糖溶液与pva水溶液混合均匀,得到cs/pva溶液;其中:所述壳聚糖溶液与pva水溶液的体积比为0.5~2:1;
(d)按比例将步骤(c)所述cs/pva溶液与步骤(b)所述w1/o乳液混合,超声分散均匀后,得到稳定的w1/o/w2乳液,其中:所述cs/pva溶液与w1/o乳液的体积比为2~6:1;
(e)取适量步骤(a)所述透明质酸钠溶液,加入pva溶液,配制has/pva溶液,其中:所述透明质酸钠溶液与步骤(c)采用的壳聚糖溶液的体积比为1:1,所述has/pva溶液与w1/o/w2乳液的体积比为4~6:1;
(f)将步骤(d)所得w1/o/w2乳液与步骤(e)所得has/pva溶液混合,室温搅拌0.5~2h后升温至45℃,继续搅拌20~40min,然后加入适量三聚磷酸钠水溶液,搅拌1~2h后静置8~12h,离心、洗涤,收集沉淀,冷冻干燥,制得所述的va@plga-cs-ha微球。
优选地,上述技术方案,步骤(a)所述plga溶液质量百分比浓度优选为10wt%;所述壳聚糖溶液的质量百分比浓度优选为1wt%;透明质酸钠溶液的质量百分比浓度优选为1.5wt%。
优选地,上述技术方案,步骤(b)所述plga溶液与va溶液的体积比优选为5:1。
优选地,上述技术方案,步骤(c)所述壳聚糖溶液与pva水溶液的体积比优选为1:1,所述pva水溶液的质量百分比浓度优选为3wt%。
优选地,上述技术方案,步骤(d)所述cs/pva溶液与w1/o乳液的体积比优选为4:1。
优选地,上述技术方案,步骤(e)所述has/pva溶液与w1/o/w2乳液的体积比为5:1,所述加入的pva溶液质量百分比浓度优选为0.4wt%。
优选地,上述技术方案,步骤(f)所述三聚磷酸钠水溶液的质量百分比浓度优选为5wt%,所述三聚磷酸钠水溶液与步骤(c)采用的壳聚糖溶液的体积比优选为1:1。
与现有技术相比,本发明涉及的一种va@plga-cs-ha/ohpmc-ha抗菌可注射水凝胶的制备方法具有如下有益效果:
(1)本发明制得的va@plga-cs-ha/ohpmc-ha抗菌可注射水凝胶具有良好力学性能、流变性能、降解性能,以及优异的溶胀性能和抗菌性能,能持续56天抑制细菌的生长,因此适合用作骨缺损修复材料。
(2)本发明方法采用的原料均具有良好的生物安全性,大大降低了临床应用的安全性风险,且本发明制备方法不涉及有机试剂或有机化学反应,反应体系温和,条件可控,不存在毒性交联剂或助剂等残留问题,整个工艺简单有效,具有很好的临床应用前景。
附图说明
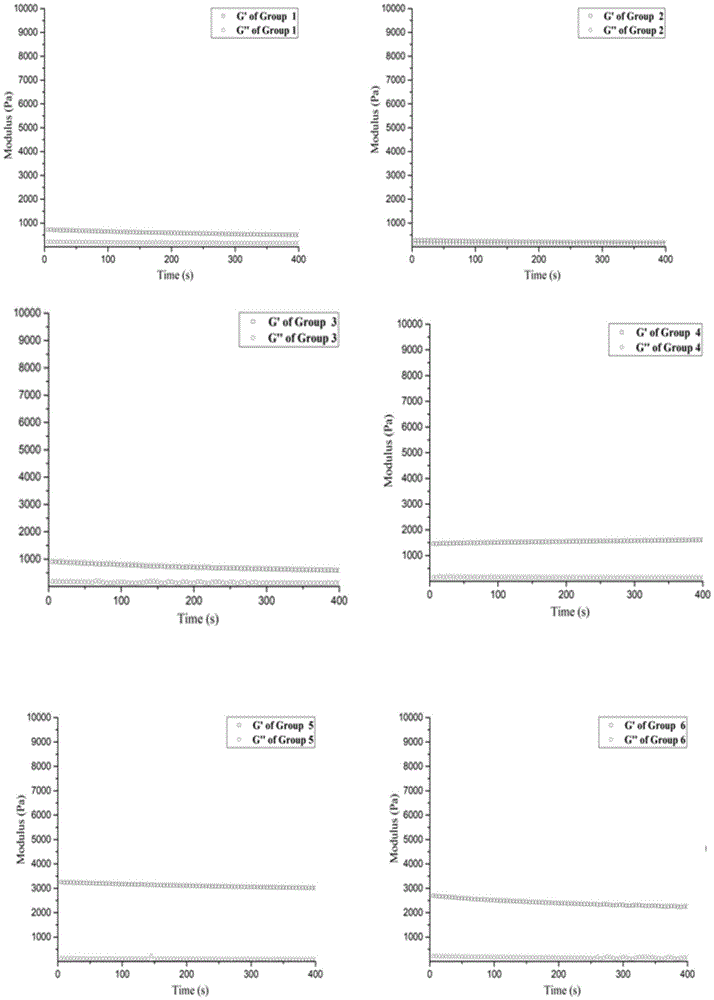
图1为实施例1~9制得的ohpmc-ha水凝胶的流变性测试结果图;(已替换)。
图2为实施例1~9制得的ohpmc-ha水凝胶的干凝胶在pbs缓冲溶液中的溶胀动力学曲线对比图;
图3为实施例10~12制得的va@plga-cs-ha/ohpmc-ha抗菌可注射水凝胶;
图4为实施例10~12制得的va@plga-cs-ha/ohpmc-ha水凝胶在pbs缓冲溶液中的溶胀动力学曲线对比图;
图5为实施例10~12制得的va@plga-cs-ha/ohpmc-ha水凝胶抗菌性能测试结果对比图;
其中:图1、图2中group1表示实施例1,group2表示实施例2,以此类推,group9表示实施例9。
具体实施方式
下面结合具体实施案例和附图对本发明作进一步详细说明。本实施案例在以本发明技术为前提下进行实施,现给出详细的实施方式和具体的操作过程来说明本发明具有创造性,但本发明的保护范围不限于以下的实施案例。
根据本申请包含的信息,对于本领域技术人员来说可以轻而易举地对本发明的精确描述进行各种改变,而不会偏离所附权利要求的精神和范围。应该理解,本发明的范围不局限于所限定的过程、性质或组分,因为这些实施方案以及其他的描述仅仅是为了示意性说明本发明的特定方面。实际上,本领域或相关领域的技术人员明显能够对本发明实施方式作出的各种改变都涵盖在所附权利要求的范围内。
为了更好地理解本发明而不是限制本发明的范围,在本申请中所用的表示用量、百分比的所有数字、以及其他数值,在所有情况下都应理解为以词语“大约”所修饰。因此,除非特别说明,否则在说明书和所附权利要求书中所列出的数字参数都是近似值,其可能会根据试图获得的理想性质的不同而加以改变。各个数字参数至少应被看作是根据所报告的有效数字和通过常规的四舍五入方法而获得的。
实施例1
本实施例的ohpmc-ha可注射水凝胶的制备方法,所述方法包括如下步骤:
(1)氧化羟丙基羧甲基纤维素(ohpmc)的制备
将羟丙基羧甲基纤维素溶解于蒸馏水中配制成质量百分比浓度为10%的羟丙基羧甲基纤维溶液,然后按体积比v羟丙基羧甲基纤维溶液:v高碘酸钠溶液=5:1加入浓度为100mg/ml的高碘酸钠溶液,避光反应24h,再加入与高碘酸钠溶液等体积的乙二醇,继续搅拌30~45min,蒸馏水透析2~3d(截留分子量:500),-50℃冷冻干燥后制得。
(2)胺化透明质酸(ha-ada)的制备
将透明质酸钠溶于蒸馏水中配制质量百分比浓度为2%的透明质酸溶液,然后按摩尔比1:1的比例加入己二酸二酰肼,待两者完全溶解后使用0.1m的hcl调节反应体系ph至4.7~4.8,再按摩尔比1:2的比例加入1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐,调节ph在4.7~4.8,室温反应12h,蒸馏水透析2~3d(截留分子量:500),-50℃冷冻干燥后制得。
(3)氧化羟丙基羧甲基纤维素-胺化透明质酸水凝胶(ohpmc-ha)的制备
将步骤(1)制得的氧化羟丙基羧甲基纤维素溶于蒸馏水中配成质量百分比浓度为1%的氧化羟丙基羧甲基纤维素溶液;将步骤(2)制得的胺化透明质酸溶于蒸馏水中配成质量百分比浓度为1%的胺化透明质酸溶液;然后将两种溶液等体积混合,漩涡10s混匀,凝胶后制得。
实施例2
本实施例的ohpmc-ha可注射水凝胶的制备方法与实施例1基本相同,区别仅在于:步骤(3)中氧化羟丙基羧甲基纤维素溶液的质量百分比浓度为1.5%,胺化透明质酸溶液的质量百分比浓度为1.5%。
实施例3
本实施例的ohpmc-ha可注射水凝胶的制备方法与实施例1基本相同,区别仅在于:步骤(3)中氧化羟丙基羧甲基纤维素溶液的质量百分比浓度为2%,胺化透明质酸溶液的质量百分比浓度为2%。
实施例4
本实施例的ohpmc-ha可注射水凝胶的制备方法与实施例1基本相同,区别仅在于:步骤(3)中氧化羟丙基羧甲基纤维素溶液的质量百分比浓度为1.5%,胺化透明质酸溶液的质量百分比浓度为1%。
实施例5
本实施例的ohpmc-ha可注射水凝胶的制备方法与实施例1基本相同,区别仅在于:步骤(3)中氧化羟丙基羧甲基纤维素溶液的质量百分比浓度为1.5%,胺化透明质酸溶液的质量百分比浓度为1.5%。
实施例6
本实施例的ohpmc-ha可注射水凝胶的制备方法与实施例1基本相同,区别仅在于:步骤(3)中氧化羟丙基羧甲基纤维素溶液的质量百分比浓度为1.5%,胺化透明质酸溶液的质量百分比浓度为2%。
实施例7
本实施例的ohpmc-ha可注射水凝胶的制备方法与实施例1基本相同,区别仅在于:步骤(3)中氧化羟丙基羧甲基纤维素溶液的质量百分比浓度为2%,胺化透明质酸溶液的质量百分比浓度为1%。
实施例8
本实施例的ohpmc-ha可注射水凝胶的制备方法与实施例1基本相同,区别仅在于:步骤(3)中氧化羟丙基羧甲基纤维素溶液的质量百分比浓度为2%,胺化透明质酸溶液的质量百分比浓度为1.5%。
实施例9
本实施例的ohpmc-ha可注射水凝胶的制备方法与实施例1基本相同,区别仅在于:步骤(3)中氧化羟丙基羧甲基纤维素溶液的质量百分比浓度为2%,胺化透明质酸溶液的质量百分比浓度为2%。
分析测试如下:
1、ohpmc-ha可注射水凝胶的成胶时间测试
用注射器分别吸取实施例1~9漩涡混匀后的混合物,注射于小玻璃瓶中。通过放倒小瓶来观察混合物状态,溶液不再流动时即为凝胶,记录时间,每组重复3次,各实施例的凝胶时间如表1所示。
表1实施例1~9的ohpmc-ha水凝胶的成胶时间对比表
由表1可以看出,可注射水凝胶的成胶时间在3~7min最为适宜(成胶时间太快不利于之后加入的va@plga-cs-ha微球的分散,成胶时间过长会导致之后加入的va@plga-cs-ha微球出现沉积)。在9组的样品中除了实施例1、实施例2、实施例3这3组之外其余各组的成胶时间均满足3~7min的要求。
2、ohpmc-ha可注射水凝胶的流变性能测试
使用流变仪分析实施例1~9制得的ohpmc-ha水凝胶的流变学。将所制备的不同组成比例的ohpmc-ha水凝胶(20.0mm直径和1.0mm厚度)放置1h后置于37℃流变仪的下板上。在1%应变下将角频率从1~150rad/s来进行频率扫描。各实施例ohpmc-ha水凝胶的流变性测试结果如图1所示。
通过旋转流变仪分析该体系水凝胶于37℃时的基本力学性能。其中,水凝胶的强度可以通过储存模量g’的大小而判断,可以看出在9组的样品中实施例9的储存模量g’最大,而实施例1~3的储存模量g’和损耗模量g”较为接近,可以判断这3组的水凝胶成粘流态,力学性能较差,不满足作为可注射水凝胶的要求。
3、ohpmc-ha可注射水凝胶的溶胀性能测试
精确称量冷冻干燥后的凝胶样品(5mm×5mm)质量,记为wd,然后置于15ml离心管中,加入10mlpbs缓冲溶液(ph=7.4)浸泡,最后置于37℃恒温摇床中以150r/min连续摇动。每隔一定时间间隔取出样品,用滤纸小心拭干样品表面水分,测量样品此时质量记为ws,则凝胶的溶胀比可由以下公式计算所得:q=ws/wd(每个样品以3个平行样为一组)。
水凝胶的溶胀动力学曲线通过各组水凝胶冷冻干燥之后的干胶在37℃pbs缓冲溶液(ph=7.4)中浸泡至溶胀平衡获得,选择不同的时间间隔以监测吸水后凝胶重量的变化。从图2中可以看到实施例4~9的水凝胶都能够快速达到溶胀平衡,大致在120min就能达至平衡,其中实施例9的溶胀比最低为3.66。而实施例1~3组的水凝胶由于吸水性过高导致在120min后水凝胶基本溃散,而与流变性测试的结果相符合。
综合上述3项测试结果可知,实施例9的水凝胶具有适宜的成胶时间,较好的力学强度和较小的溶胀比,在9组的水凝胶综合性能较好,所以在后续实施例10~12va@plga-cs-ha/ohpmc-ha抗菌水凝胶的制备中,ohpmc和ha-ada的浓度和比例均按采用实施例9制备水凝胶采用的原料比例。
实施例10
本实施例的一种va@plga-cs-ha/ohpmc-ha抗菌可注射水凝胶的制备方法,所述方法包括如下步骤:
(1)将氧化羟丙基羧甲基纤维素(ohpmc)溶解于蒸馏水中,配制质量百分比浓度为2wt%的ohpmc溶液;将胺化透明质酸(ha-ada)溶解于蒸馏水中,配制质量百分比浓度为2wt%的ha-ada溶液;然后将ohpmc溶液和ha-ada溶液等体积混合均匀,得到ohpmc-ha均匀混合液;
(2)按va@plga-cs-ha微球与ohpmc-ha水凝胶的用量比为0.1g:1ml的比例将va@plga-cs-ha微球加入到步骤(1)制得的ohpmc-ha均匀混合液中,混匀,制得本发明所述的va@plga-cs-ha/ohpmc-ha抗菌可注射水凝胶,命名为va@plga-cs-ha/ohpmc-ha01;
其中:
步骤(2)所述va@plga-cs-ha微球采用下述方法制得,所述方法包括如下步骤:
(a)将plga溶解于二氯甲烷中,配制成质量百分比浓度为10wt%的plga溶液,备用;将壳聚糖溶解于质量百分比浓度为2wt%的冰乙酸中,配制成质量百分比浓度为1wt%的壳聚糖溶液,备用;将透明质酸钠溶解于蒸馏水中,配制质量百分比浓度为1.5wt%的透明质酸钠溶液,备用;将万古霉素盐酸盐(va)溶解于蒸馏水中,配制质量百分比浓度为50mg/ml的万古霉素溶液,备用;将聚乙烯醇(pva)溶解于蒸馏水中,配制质量百分比浓度为3wt%的pva溶液,备用;将三聚磷酸钠溶解于蒸馏水中配制成质量百分比浓度为5wt%的三聚磷酸钠溶液,备用;
(b)按体积比vplga:vva=5:1,将plga溶液与va溶液混合,漩涡混均5min,然后超声5min,形成w1/o乳液;
(c)按体积比vcs:vpva=1:1将壳聚糖溶液和pva溶液混合均匀,得cs/pva溶液;
(d)按体积比vcs/pva:vw1/o=4:1,将cs/pva溶液与w1/o乳液混合,漩涡混均5min,超声5min,重复2~3次,得到稳定的w1/o/w2乳液;
(e)量取与步骤(3)中所用的壳聚糖溶液体积相当的透明质酸钠溶液,然后用加入质量百分比浓度为0.4wt%的pva溶液,配制成has/pva溶液,has/pva溶液的总体积为w1/o/w2乳液的体积的5倍;
(f)将步骤(4)所得w1/o/w2乳液与步骤(e)所得has/pva溶液混合,室温下800rpm机械搅拌1h,然后在1000rpm下升温至45℃,搅拌30min,然后再800rpm条件下加入与步骤(3)中所用的壳聚糖溶液体积相当的三聚磷酸钠溶液,继续搅拌1.5h,静置过夜(12h),8000rpm离心5min,蒸馏水洗涤分散,8000rpm离心,收集沉淀,冷冻干燥,制得va@plga/cs/ha-50复合微球。
实施例11
本实施例的一种va@plga-cs-ha/ohpmc-ha抗菌可注射水凝胶的制备方法,与实施例1基本相同,区别仅在于:本实施例步骤(2)中va@plga-cs-ha微球与ohpmc-ha水凝胶的用量比为0.2g:1ml,命名为va@plga-cs-ha/ohpmc-ha02。
实施例12
本实施例的一种va@plga-cs-ha/ohpmc-ha抗菌可注射水凝胶的制备方法,与实施例1基本相同,区别仅在于:本实施例步骤(2)中va@plga-cs-ha微球与ohpmc-ha水凝胶的用量比为0.3g:1ml,命名为va@plga-cs-ha/ohpmc-ha03。
性能测试及表征
1、将上述实施例10~12的va@plga-cs-ha/ohpmc-ha抗菌可注射水凝胶的成胶时间测试的成胶时间进行测试,测试结果如表2所示。
表2实施例10~12的va@plga-cs-ha/ohpmc-ha水凝胶的成胶时间对比表
含不同va@plga-cs-ha微球的va@plga-cs-ha/ohpmc-ha抗菌可注射水凝胶的成胶时间如表2所示,可以看出va@plga-cs-ha微球的加入对水凝胶的成胶时间有轻微的影响,水凝胶中所含的微球的量越多,成胶时间会有所增加,但整体影响不大。
2、va@plga-cs-ha/ohpmc-ha抗菌可注射水凝胶的流变性能测试
分别将上述实施例10~12制得的产物va@plga-cs-ha/ohpmc-ha01、va@plga-cs-ha/ohpmc-ha02、va@plga-cs-ha/ohpmc-ha03进行流变性能测试,测试结果如图3所示。由图3可以看出,微球的添加可以使水凝胶的力学性能有所增加,而且随着微球的添加量的增多,水凝胶的存储模量有明显提高。
3、va@plga-cs-ha/ohpmc-ha抗菌可注射水凝胶的溶胀性能测试
图4为实施例10~12制得的va@plga-cs-ha/ohpmc-ha水凝胶在pbs缓冲溶液中的溶胀动力学曲线对比图;从图4可以看出va@plga-cs-ha微球的加入可以使水凝胶的溶胀比有轻微的下降,但是总体来说对ohpmc-ha水凝胶的溶胀性能影响不大。
4、va@plga-cs-ha/ohpmc-ha抗菌可注射水凝胶的抗菌性能测试
抗菌性能测试方法如下:
(1)配制lb培养液和生理盐水,对金黄葡萄球菌进行复苏,并且用生理盐水将菌液稀释至菌液浓度为105cfu/ml。
(2)称取50mg水凝胶样品,紫外灭菌处理,将其放入50ml的锥形瓶中,加入1ml去lb培养液,并加入0.2ml菌液,在37℃摇床120rpm的条件下将样品和菌液共培养(共培养时间点分别为:1d,3d,7d,14d,21d,28d,35d,42d,56d)。
(3)取100μl的混合液采用培养基倾注法接种平皿,将培养皿置于37℃细菌培养箱培养24h后计算菌落数。计算抑菌率,计算公式如下:
relativecfu%=cfu(24h)/cfu(0h)×100%。
图5为含有不同量的va@plga-cs-ha微球的va@plga-cs-ha/ohpmc-ha水凝胶抗菌性能测试结果,可以看出3组水凝胶在第7天时杀菌率均达到90%,在第14天时3组的抑菌率均接近99%,而在测试的后期可以看出3组的relativecfu有轻微的上升,但3组仍能有效抑制细菌的生长(在56天时3组的relativecfu仍小于1%)。
- 还没有人留言评论。精彩留言会获得点赞!