2-丙酰基-1-吡咯啉的合成方法与流程
本发明属于化学合成技术领域,具体涉及2-丙酰基-1-吡咯啉的合成方法。
背景技术:
2-丙酰基-1-吡咯啉作为一种美拉德香料,其用途十分广泛,常用作为烘培类、饮料、糖果、香稻等食品的添加剂。2-丙酰基-1-吡咯啉比同类香料用途更为广泛,香气更加浓郁,但是由于2-丙酰基-1-吡咯啉的价格更为昂贵,因此一条经济实惠、产率高的合成路线尤为重要。目前合成2-丙酰基-1-吡咯啉的主要方法有:1.由2-丙酰基吡咯在金属催化作用下还原最后在碳酸银作用下合成2-丙酰基-1-吡咯啉。2.由脯氨酸一类通过酯化、氯化、水解最终得到2-丙酰基-1-吡咯啉。但这些合成方法都有着以下缺点(i)整个合成过程的成本高;(ii)反应所需时间长、效率低;(iii)现有合成工艺中需要使用危险/有毒的试剂,例如nacn,hcn和次氯酸叔丁酯或叔丁基锂,并且最后阶段中酸性或碱性的处理被证明对总产率不利。
技术实现要素:
本发明的目的在于提供一种2-丙酰基-1-吡咯啉的合成方法,本发明合成方法简单,合成产率高,具有更高的经济价值。
为了达到上述目的,本发明所采取的技术方案是:
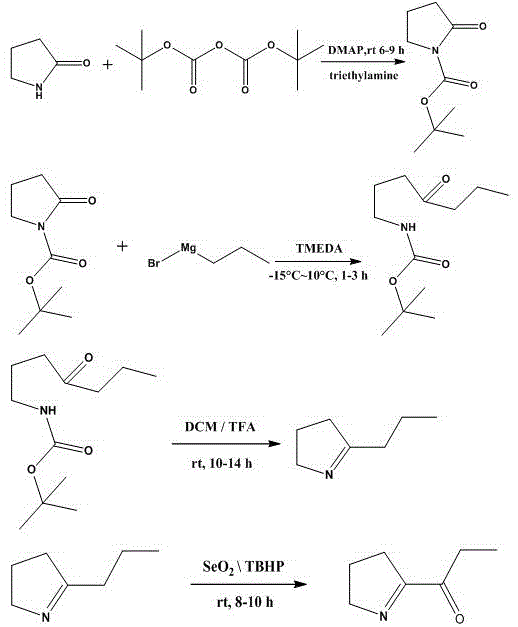
2-丙酰基-1-吡咯啉的合成方法,包括以下步骤:
1)将2-吡咯烷酮溶解于二氯甲烷,待其完全溶解,再依次加入4-二甲氨基吡啶dmap和三乙胺;最后缓慢加入二碳酸二叔丁酯,进行加成反应;反应结束后,用蒸馏水与饱和食盐水多次洗涤反应液,收集有机相;进行柱层析,洗脱剂为石油醚/乙酸乙酯,得到纯n-boc-吡咯烷酮;
2)将n-boc-吡咯烷酮溶于四氢呋喃,在低温环境下滴加四甲基乙二胺tmeda和格氏试剂,进行格氏反应;反应停止,加入盐酸溶液直至反应液不再产生气泡,随后用有机溶剂进行萃取,水溶液进行多次洗涤,加入干燥剂干燥脱水,得到纯n-boc-4-氧代-庚胺;
3)利用三氟乙酸与二氯甲烷的混合溶液,与n-boc-4-氧代-庚胺发生消去反应;用碱性溶液调ph为8-10,有机溶剂萃取,经过多次酸洗和碱洗,用有机溶剂萃取,最后加入干燥剂干燥脱水,得到纯丙基吡咯啉;
4)向二氧化硒中加入,过氧化叔丁醇的二氯甲烷溶液,搅拌30分钟,随后滴加丙基吡咯啉的二氯甲烷溶液,发生氧化反应;反应停止,大量水洗,加入干燥剂干燥脱水,柱层析洗脱得到纯产物2-丙酰基-1-吡咯啉。
步骤1)中,2-吡咯烷酮与二碳酸二叔丁酯摩尔比为1:(1-1.5);2-吡咯烷酮与4-二甲氨基吡啶和三乙胺的摩尔比为2:1:3;步骤1)中加成反应的温度为20℃~30℃,反应时间6~9小时。
步骤2)和步骤3)和步骤4)中所述的干燥剂为无水硫酸钙、无水硫酸镁、无水氯化钙、无水硫酸钠中的至少一种。
步骤2)和步骤3)中所述的有机溶剂为乙醚、二氯甲烷、甲苯、三氯甲烷中的一种。
步骤2)中,n-boc-吡咯烷酮与格氏试剂摩尔比为1:(1-1.2);n-boc-吡咯烷酮与四甲基乙二胺摩尔比为1:1.2;步骤2)中格氏反应的温度为-15℃~-10℃,反应时间1~3小时。
步骤3)中消去反应的温度为20℃~30℃,反应时间10~14小时。
步骤3)中所述的碱性溶液为氢氧化钠、氢氧化钾中的一种;碱性溶液浓度为1~2m;酸溶液为1-3mol/l的盐酸溶液。
步骤3)中,n-boc-4-氧代-庚胺与三氟乙酸摩尔比为1:(4-5);步骤3)中,二氯甲烷与三氟乙酸体积比为1:(3-5)。
步骤4)中氧化反应的温度为20℃~30℃,反应时间8~10小时。
步骤4)中,二氧化硒与丙基吡咯啉的摩尔比为1:5;二氧化硒与过氧化叔丁醇摩尔比为1:5。
本发明有益的效果是:
本发明的合成方法相比较之前的其他合成路线操作更为简单。通过从反应温度、投料比、反应时间和产率四个方面来看,整个合成路线节约成本并缩短时间,相对于其他路线需要极低温度(约-80℃)下进行反应,本发明极大地改善了反应温度的条件。最后一步利用里利氧化法将丙基吡咯啉氧化得到2-丙酰基-1-吡咯啉,里利氧化法温度需要100℃,而本发明只需要在室温下反应,并且通过改变氧化剂的投料量,就可以将反应产率从30%提升到55%。纵观整个合成路线,本发明仅需约2天的时间就可以合成目标产物,而其他合成方法则需要4-5天,因此本发明更加适用于工业化生产。
附图说明
图1本发明合成路线图。
具体实施方式
下面将结合实施例对本发明的实施方案进行详细描述,本领域技术人员将会理解,以下实施例仅为本发明的优选实施例,以便更好地理解本发明,因而不应视为限定本发明的范围。
实施例一:
将5g2-吡咯烷酮(58.75mmol)溶于100ml二氯甲烷溶液中,待其完全溶解,再依次加入0.75g4-二甲氨基吡啶(dmap4.876mmol)、12.5ml三乙胺(88mmol)。最后缓慢加入13.97g二碳酸二叔丁酯(15ml64mmol),在滴加过程中产生大量气泡,溶液颜色逐渐由微黄变成金黄。反应9小时后,用20ml蒸馏水洗涤反应液,将下层有机相用饱和食盐水(nacl20ml)洗涤2次。收集下层有机相,减压旋蒸得到黄色液体的粗产物12.46g。将粗产物进行柱层析,洗脱剂石油醚/乙酸乙酯(1:1,v/v),最终得到纯n-boc-吡咯烷酮10.19g。
将丙基溴化镁在-10℃的环境中,搅拌状态下滴加四甲基乙二胺tmeda(10ml),再缓慢加入n-boc-吡咯烷酮(10.19g,55.0mmol)的thf(40ml)溶液。在-10℃环境中,反应2小时,用2m的hcl(100ml)溶液淬灭反应,并用et2o进行萃取(3×60ml),合并的有机相用饱和食盐水(2×60ml)和饱和nahco3溶液(60ml)洗涤,用无水na2so4干燥,减压蒸馏得到n-boc-4-氧代-庚胺。
在0℃氛围下,往n-boc-4-氧代-庚胺11.33g(49.5mmol)中滴加dcm/tfa(75ml,8:2)的混合溶剂。随后在室温下搅拌12小时,将反应混合物冷却至0℃,并用足量的naoh(2m)调节ph为10。然后,用et2o(2×100ml)萃取反应液,将合并的有机相用1mhcl(3×100ml)洗涤,收集水相。用et2o(50ml)洗涤合并的水溶液,收集水相用2mnaoh调节ph为10,然后用et2o(4×75ml)萃取。有机相用盐水(2×50ml)洗涤,用na2so4干燥。通过减压蒸馏除去溶剂,得到淡黄色液体丙基吡咯啉(4.28g38.61mmol)。
称取0.856gseo2(7.722mmol)加入8.5mlch2cl2,再加入19ml2m过氧化叔丁醇tbhp的ch2cl2溶液,反应半小时。向上述反应液中滴加4.28g丙基吡咯啉在10mlch2cl2溶液,搅拌9小时反应液颜色逐渐加深。反应结束加入2g的碳粉吸附胶体硒并过滤。随后加入大量的水进行水洗,用na2so4干燥,减压旋蒸到粗产物。将粗产物进行柱层析,洗脱剂为石油醚/乙醚梯度淋洗(体积比:9:1→3:1→1:1),得到黄色液体纯产物2-丙酰基-1-吡咯啉2.5g(20mmol),经纯度检验为99%。1hnmr(400mhz,cdcl3)δ4.10(2h,tt,j=7.5,2.4hz,h-c(5)),2.92(2h,q,j=7.3hz,ch2ch3),2.73(2h,tt,j=8.4,2.3hz,h-c(3)),1.93(2h,m,h-c(4)),1.12(3h,t,j=7.4hz,ch3);13cnmr(100mhz,cdcl3)δ203.4,177.0,65.5,36.4,36.3,24.9,10.5。
以上所述仅为本发明的较佳实施例,凡依本发明申请专利范围所做的均等变化与修饰,皆应属本发明的涵盖范围。
- 还没有人留言评论。精彩留言会获得点赞!