异相碱性催化剂及基于该异相碱性催化剂的α,β-不饱和化合物的连续流制备方法与流程
本发明涉及一种有机催化合成技术领域,特别是涉及异相碱性催化剂及基于该异相碱性催化剂的α,β-不饱和化合物的连续流制备方法。
背景技术:
α,β-不饱和化合物的合成反应是一种重要的增长碳链和引入不饱和碳碳双键的方法,在药物、天然产物、精细化学品以及杂环化合物等高附加产品的有机合成方面应用非常广泛。因此,α,β-不饱和化合物的新合成方法研究具有重要的实用价值,受到很多相关领域科研工作者的关注。
传统的α,β-不饱和化合物的合成方法通常是采用批次反应技术、以cdi2、吡啶、羟基磷灰石等作为催化剂来进行α,β-不饱和化合物的催化合成。但是,传统的合成方法存在很多局限性,比如催化剂可回收性差、难分离、对环境不友好,以及产物后处理麻烦、纯度低、较难快速大批量生产等。
技术实现要素:
有鉴于此,本发明的目的在于提供一种易于回收、具有高催化活性和稳定性、对环境友好的异相碱性催化剂。
本发明的目的在于提供一种可提高产品的纯度,降低了生产成本,易于大批量生产的基于异相碱性催化剂的α,β-不饱和化合物的连续流制备方法。
为了达成上述目的,本发明的解决方案是:
异相碱性催化剂的制备方法,包括如下步骤:
步骤a、将市售氯球、含氮化合物和溶剂置于反应容器中,并加入碳酸氢钠调节ph至碱性,使其在温度为65~70℃的碱性环境中反应10~12小时;
步骤b、反应结束后,往步骤a中的反应容器中加入蒸馏水溶解固体杂质,抽滤得滤渣,然后用乙醇淋洗后干燥得到异相碱性催化剂,
其中,步骤a中,市售氯球中氯的质量分数为18~19%,市售氯球中的氯与含氮化合物的摩尔比为1∶1~1∶1.2,市售氯球中的氯与碳酸氢钠的摩尔比为1∶1.3~1∶1.5。
进一步地,步骤a中,所用溶剂为n,n-二甲基甲酰胺(dmf)。
进一步地,步骤a中,所用含氮化合物为咪唑、二乙烯三胺、精胺、三乙烯四胺、1,6-己二胺或者盐酸胍。
进一步地,步骤b中,所得异相碱性催化剂的结构式如下所示:
采用上述技术方案后,本发明一种异相碱性催化剂的制备方法,具有以下有益效果:将氯球、含氮化合物和溶剂置于反应容器中,并调节ph至碱性环境中反应12小时。反应结束后,干燥得到异相碱性催化剂。使用时通过简单洗涤、干燥即可回收再使用,经实验测试,催化剂表现出高稳定性和催化活性,并且对环境友好。
一种基于所述的异相碱性催化剂的α,β-不饱和化合物的连续流制备方法,包括以下步骤:
步骤1、将异相碱性催化剂填充到反应器中,反应器与泵相连接;
步骤2、将苯甲醛及其衍生物、丙二腈及其衍生物和溶剂于一反应容器中混合,形成反应液,反应容器与上述泵相连接,反应液在泵中流速为0.25ml/min的室温下流反应40min;
步骤3、将步骤2中流反应生成的溶液进行旋转蒸发操作以获得粗产品,然后经过乙醇重结晶、干燥得到目标产物α,β-不饱和化合物;
其中,步骤1中,异相碱性催化剂的用量为1.0g,步骤2中,苯甲醛及其衍生物和丙二腈及其衍生物的摩尔比为1∶1.2。
进一步地,步骤1中,所用异相碱性催化剂为1,6-己二胺功能化氯球催化剂。
进一步地,步骤2中,所用溶剂为纯乙酸乙酯或体积比为25∶1的乙醇/dmf。
进一步地,步骤2中,苯甲醛及其衍生物的结构式为:
进一步地,步骤2中,丙二腈及其衍生物为丙二腈或者丙二腈衍生物,其中丙二腈衍生物的结构式为
进一步地,步骤3所得的α,β-不饱和化合物的结构式为
采用上述技术方案后,本发明一种基于异相碱性催化剂的α,β-不饱和化合物的连续流制备方法,具有以下有益效果:将α,β-不饱和化合物的合成与流技术结合起来,产品通过重结晶纯化,后处理简单且对处理过程不需使用大量有机溶剂进行柱色谱分离,环境友好;流技术的运用,提高了产品的纯度、降低了生产成本以及易于α,β-不饱和化合物的快速大批量生产。
附图说明
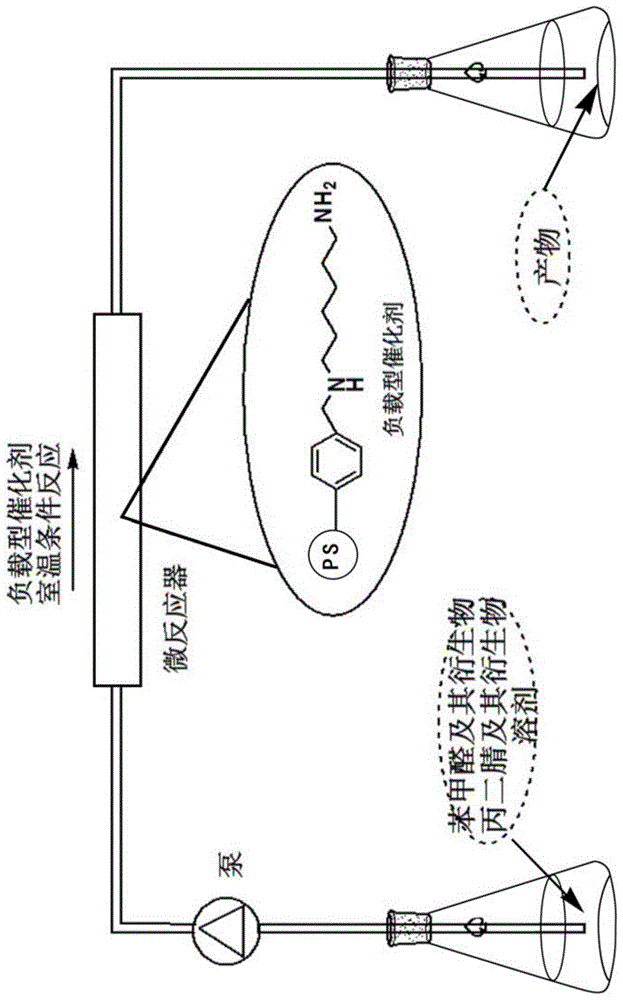
图1为本发明中α,β-不饱和化合物的制备简图。
具体实施方式
为了进一步解释本发明的技术方案,下面通过具体实施例来对本发明进行详细阐述。
一、异相碱性催化剂的制备
步骤a、将市售ps氯球(交联度为4%,含氯量为18~19%)、含氮化合物和溶剂置于反应容器中,并加入一定量的nahco3调节ph至碱性,使其在温度为70℃的碱性环境中反应12小时。
步骤b、反应结束后,往步骤1中的反应瓶中加入适量蒸馏水溶解固体杂质,抽滤得滤渣,然后用乙醇淋洗3次、50℃下干燥后,得到异相碱性催化剂。
其中,步骤a中,市售氯球中氯的质量分数为18%-19%,氯球中的氯与含氮化合物的摩尔比为1∶1.2,市售氯球中的氯与碳酸氢钠的摩尔比为1∶1.3~1∶1.5,市售氯球的质量与溶剂的体积比为5∶1ml/g。
步骤a中,溶剂为n,n-二甲基甲酰胺(dmf)。
步骤a中,所用含氮化合物为有机胺类化合物,含氮化合物的结构式如下式i-式vi(共6种)任一式所示:
步骤b中,所得异相碱性催化剂的结构式如下式vii-式xii(共6种)任一式所示:
本发明异相碱性催化剂制备方法的制备方法如下:
采用上述技术方案后,通过本发明一种异相碱性催化剂的制备方法制得的催化剂,将市售氯球、相应的含氮化合物(盐酸胍、二乙烯三胺、三乙烯四胺、精胺、1,6-己二胺或咪唑)和溶剂置于反应容器中反应,最终得到6种相应的胺功能化氯球催化剂。相较于传统的以cdi2、吡啶、羟基磷灰石等为催化剂来批次反应生成α,β-不饱和化合物的合成方法,本发明的催化剂用于α,β-不饱和化合物的制备时,不仅易于回收、具有高催化活性(可缩短下述α,β-不饱和化合物制备的反应时间至40min,收率为97%)和稳定性(循环使用10次时,下述α,β-不饱和化合物制备产率仍在80%以上),并且对环境友好。
二、α,β-不饱和化合物制备
步骤1、将异相碱性催化剂填充到一个反应器中,反应器优选为柱反应管,柱反应管连接有输送管路,输送管路上安装有泵,即柱反应管与泵相连通连接;
步骤2、将苯甲醛及其衍生物、丙二腈及其衍生物和溶剂于一反应容器中混合,形成反应液,反应容器与上述输送管路相连通连接,即反应容器与泵相连通连接,反应液在泵中流速为0.25ml/min的室温下流反应40min;
步骤3、将步骤2中流反应生成的溶液进行旋转蒸发操作以获得粗产品,然后经过乙醇重结晶、干燥得到目标产物α,β-不饱和化合物。
其中,步骤1中,异相碱性催化剂优选为ps-dah,其用量为1.0g;步骤2中,所述溶剂为体积比为25∶1的乙醇/dmf或乙酸乙酯,并且苯甲醛及其衍生物和丙二腈及其衍生物的摩尔比为1∶1.2,物质的量分别为0.006mol和0.0072mol,苯甲醛及其衍生物、丙二腈及其衍生物和溶剂的总体积为10ml。
步骤2中,苯甲醛及其衍生物的结构式为
步骤2中,丙二腈衍生物的结构式如式xiv所示:
其中,r3为氰基、酯基。
步骤3制得的α,β-不饱和化合物的结构式如式xv的其中一种所示:
其中,r1为氢、羟基;
r2为氢、羟基、卤素、甲氧基、硝基;
r3为氰基、酯基。
本发明α,β-不饱和化合物制备简图如图1所示,流技术的使用提高了产品的纯度、降低了生产成本以及易于α,β-不饱和化合物的快速大批量生产。
下面α,β-不饱和化合物制备以异相碱性催化剂采用ps-dah为例进行说明:
实施例1
苄烯丙二腈的制备(结构式如下):
制备方法:将ps-dah(1.0g)填充到一个柱反应管中,并将柱反应管与泵相连接。然后,将0.006mol的苯甲醛和0.0072mol的丙二腈加入到10ml的量筒(即反应容器)中,然后将体积比为25∶1的乙醇/dmf溶剂滴加到量筒中,直到量筒中反应液的体积为10ml。将10ml反应液转移到柱反应管中,并将泵中的流速设置为0.25ml/min,反应液在室温下流反应40min。反应结束后,将生成液进行旋转蒸发操作以获得粗产品,然后经过乙醇重结晶、干燥得到0.90g目标产物苄烯丙二腈,收率为97%。
与传统方法制备的化合物相比,该路线所得产品纯度更高,熔程更短。相关表征数据如下:m.p.83.3℃;ft-ir(kbrdisc)cm-1:2222.6,1590.5,1265.6,1217.4,1100.69;1hnmr(500mhz,cdcl3)δ:7.93(d,j=7.2hz,2h),7.81(s,1h),7.66(t,j=7.5hz,1h),7.57(t,j=7.7hz,2h).
实施例2
a-环甲基乙酸乙酯的制备(结构式如下):
制备方法:将ps-dah(1.0g)填充到一个柱反应管中,并将柱反应管与泵相连。然后,将0.006mol的苯甲醛和0.0072mol的氰乙酸乙酯加入到10ml的量筒中,然后将体积比为25∶1的乙醇/dmf溶剂滴加到量筒中,直到量筒中反应液的体积为10ml。将10ml反应液转移到柱反应管中,并将泵中的流速设置为0.25ml/min,反应液在室温下流反应40min。反应结束后,将生成液进行旋转蒸发操作以获得粗产品,然后经过乙醇重结晶、干燥得到1.04g目标产物a-环甲基乙酸乙酯,收率为86%。
与传统方法制备的化合物相比,该路线所得产品纯度更高,熔程更短。该化合物的表征结果如下:m.p.50.2℃;ft-ir(kbrdisc)cm-1:2987.2,2223.1,1723.6,1573.2;1hnmr(500mhz,cdcl3)δ:8.28(s,1h),8.02(d,j=6.9hz,2h),7.63–7.55(m,1h),7.53(d,j=8.3,6.7hz,2h),4.41(q,j=7.1hz,2h),1.43(t,j=7.1hz,3h).
实施例3
2-氰基-3-(4-羟基苯基)丙烯酸乙酯的制备(结构式如下):
制备方法:将ps-dah(1.0g)填充到一个柱反应管中,并将柱反应管与泵相连接。然后,将0.006mol的4-羟基苯甲醛和0.0072mol的氰乙酸乙酯加入到10ml的量筒中,然后将体积比为25∶1的乙醇/dmf溶剂滴加到量筒中,直到量筒中反应液的体积为10ml。将10ml反应液转移到柱反应管中,并将泵中的流速设置为0.25ml/min,反应液在室温下流反应40min。反应结束后,将生成液进行旋转蒸发操作以获得粗产品,然后经过乙醇重结晶、干燥得到1.17g目标产物2-氰基-3-(4-羟基苯基)丙烯酸乙酯,收率为90%。
与传统方法制备的化合物相比,该路线所得产品纯度更高,熔程更短。该化合物的表征结果如下:m.p.172.3–172.5℃;ft-ir(kbrdisc)cm-1:3313.6,2227.4,1609.8,1173.0,1013.9;1hnmr(500mhz,cdcl3)δ:8.21(s,1h),7.98(d,j=8.1hz,2h),6.99(d,j=8.1hz,2h),4.40(q,j=7.1hz,2h),1.42(t,j=7.1hz,3h).
实施例4
2-(4-溴亚苄基)丙二腈的制备(结构式如下):
制备方法:将ps-dah(1.0g)填充到一个柱反应管中,并将柱反应管与泵相连。然后,将0.006mol的4-溴苯甲醛和0.0072mol的丙二腈加入到10ml的量筒中,然后将体积比为25∶1的乙醇/dmf溶剂滴加到量筒中,直到量筒中反应液的体积为10ml。将10ml反应液转移到柱反应管中,并将泵中的流速设置为0.25ml/min,反应液在室温下流反应40min。反应结束后,将生成液进行旋转蒸发操作以获得粗产品,然后经过乙醇重结晶、干燥得到1.20g目标产物2-(4-溴亚苄基)丙二腈,收率为86%。
与传统方法制备的化合物相比,该路线所得产品纯度更高,熔程更短。该化合物的表征结果如下:m.p.161.5–161.6℃;ft-ir(kbrdisc)cm-1:2227.4,1554.3;1hnmr(500mhz,cdcl3)δ:7.79(d,j=8.6hz,2h),7.75(s,1h),7.71(d,j=8.6hz,2h).
实施例5
3-(4-溴苯基)-2-氰基丙烯酸乙酯的制备(结构式如下):
制备方法:将ps-dah(1.0g)填充到一个柱反应管中,并将柱反应管与泵相连。然后,将0.006mol的4-溴苯甲醛和0.0072mol的丙二腈加入到10ml的量筒中,然后将乙酸乙酯滴加到量筒中,直到量筒中反应液的体积为10ml。将10ml反应液转移到柱反应管中,并将泵中的流速设置为0.25ml/min,反应液在室温下流反应40min。反应结束后,将生成液进行旋转蒸发操作以获得粗产品,然后经过乙醇重结晶、干燥得到1.53g目标产物3-(4-溴苯基)-2-氰基丙烯酸乙酯,收率为91%。
与传统方法制备的化合物相比,该路线所得产品纯度更高,熔程更短。该化合物的表征结果如下:m.p.96.6–97.4℃;ft-ir(kbrdisc)cm-1:1720.7,1609.3;1hnmr(500mhz,cdcl3)δ:8.20(s,1h),7.87(d,2h),7.67(d,2h),4.41(q,j=7.2hz,2h),1.42(t,j=7.2hz,3h).
实施例6
(4-甲氧基苄烯)丙二腈的制备(结构式如下):
制备方法:将ps-dah(1.0g)填充到一个柱反应管中,并将柱反应管与泵相连。然后,将0.006mol的4-甲氧基苯甲醛和0.0072mol的丙二腈加入到10ml的量筒中,然后将乙酸乙酯滴加到量筒中,直到量筒中反应液的体积为10ml。将10ml反应液转移到柱反应管中,并将泵中的流速设置为0.25ml/min,在室温下流反应40min。反应结束后,将生成液进行旋转蒸发操作以获得粗产品,然后经过乙醇重结晶、干燥得到0.96g目标产物(4-甲氧基苄烯)丙二腈,收率为87%。
与传统方法制备的化合物相比,该路线所得产品纯度更高,熔程更短。该化合物的表征结果如下:m.p.114.6–115.0℃;ft-ir(kbrdisc)cm-1:2219.2,1561.1;1hnmr(500mhz,cdcl3)δ:7.93(d,j=8.4hz,2h),7.68(s,1h),7.04(d,j=8.4hz,2h),3.94(s,3h).
实施例7
2-氰基-3-(4-甲氧基苯基)丙烯酸乙酯的制备(结构式如下):
制备方法:将ps-dah(1.0g)填充到一个柱反应管中,并将柱反应管与泵相连。然后,将0.006mol的4-甲氧基苯甲醛和0.0072mol的氰乙酸乙酯加入到10ml的量筒中,然后将体积比为25∶1的乙醇/dmf溶剂滴加到量筒中,直到量筒中反应液的体积为10ml。将10ml反应液转移到柱反应管中,并将泵中的流速设置为0.25ml/min,反应液在室温下流反应40min。反应结束后,将生成液进行旋转蒸发操作以获得粗产品,然后经过乙醇重结晶、干燥得到1.25g目标产物2-氰基-3-(4-甲氧基苯基)丙烯酸乙酯,收率为90%。
与传统方法制备的化合物相比,该路线所得产品纯度更高,熔程更短。该化合物的表征结果如下:m.p.82.1-83.4℃;ft-ir(kbrdisc)cm-1:2981.9,2938.1,2842.1,2220.7,1715.9,1585.7,1256.4,1123.8;1hnmr(500mhz,cdcl3)δ:8.19(d,j=1.8hz,1h),8.02(s,2h),7.01(d,j=7.1hz,2h),4.38(q,j=7.1,1.3hz,2h),3.91(s,3h),1.41(t,j=7.2,1.2hz,3h).
实施例8
1,1-二氰基-2-(4-硝基苯基)-乙烯的制备(结构式如下):
制备方法:将ps-dah(1.0g)填充到一个柱反应管中,并将柱反应管与泵相连。然后,将0.006mol的4-硝基苯甲醛和0.0072mol的丙二腈加入到10ml的量筒中,然后将体积比为25∶1的乙醇/dmf溶剂滴加到量筒中,直到量筒中反应液的体积为10ml。将10ml反应液转移到柱反应管中,并将泵中的流速设置为0.25ml/min,反应液在室温下流反应40min。反应结束后,将生成液进行旋转蒸发操作以获得粗产品,然后经过乙醇重结晶、干燥得到1.06g目标产物1,1-二氰基-2-(4-硝基苯基)-乙烯,收率为89%。
与传统方法制备的化合物相比,该路线所得产品纯度更高,熔程更短。该化合物的表征结果如下:m.p.159.1-159.4℃;ft-ir(kbrdisc)cm-1:2231.3,1579.9;1hnmr(500mhz,cdcl3)δ:8.41(d,j=8.6hz,2h),8.10(d,j=9.4hz,2h),7.90(s,1h).
实施例9
2-氰基-3-(4-硝基苯基)丙烯酸乙酯的制备(结构式如下):
制备方法:将ps-dah(1.0g)填充到一个柱反应管中,并将柱反应管与泵相连。然后,将0.006mol的4-硝基苯甲醛和0.0072mol的氰乙酸乙酯加入到10ml的量筒中,然后将体积比为25∶1的乙醇/dmf溶剂滴加到量筒中,直到量筒中反应液的体积为10ml。将10ml反应液转移到柱反应管中,并将泵中的流速设置为0.25ml/min,反应液在室温下流反应40min。反应结束后,将生成液进行旋转蒸发操作以获得粗产品,然后经过乙醇重结晶、干燥得到1.30g目标产物2-氰基-3-(4-硝基苯基)丙烯酸乙酯,收率为88%。
与传统方法制备的化合物相比,该路线所得产品纯度更高,熔程更短。该化合物的表征结果如下:m.p.170.4-170.8℃;ft-ir(kbrdisc)cm-1:2921.7,2226.5,1718.8,1616.6,1512.4,1267.5,122.4,858.7;1hnmr(500mhz,cdcl3)δ:8.38(d,j=8.6hz,2h),8.33(s,1h),8.16(d,j=9.2hz,2h),4.45(q,j=7.1hz,2h),1.45(t,j=7.1hz,3h).
实施例10
(2-呋喃亚甲基)丙二腈的制备(结构式如下):
制备方法:将ps-dah(1.0g)填充到一个柱反应管中,并将柱反应管与泵相连。然后,将0.006mol的糠醛和0.0072mol的丙二腈加入到10ml的量筒中,然后将体积比为25∶1的乙醇/dmf溶剂滴加到量筒中,直到量筒中反应液的体积为10ml。将10ml反应液转移到柱反应管中,并将泵中的流速设置为0.25ml/min,反应液在室温下流反应40min。反应结束后,将生成液进行旋转蒸发操作以获得粗产品,然后经过乙醇重结晶、干燥得到0.80g目标产物(2-呋喃亚甲基)丙二腈,收率为92%。
与传统方法制备的化合物相比,该路线所得产品纯度更高,熔程更短。该化合物的表征结果如下:m.p.70.8–71.4℃;ft-ir(kbrdisc)cm-1:3037.4,2219.2,1681.7,1607.9,1456.0,1397.2,1072.4,763.2;1hnmr(500mhz,cdcl3)δ:7.83(d,j=1.6hz,1h),7.54(s,1h),7.39(d,j=3.8hz,1h),6.74(dd,j=3.8,1.7hz,1h).
实施例11
2-氰基-3-(呋喃-3-基)丙烯酸乙酯的制备(结构式如下):
制备方法:将ps-dah(1.0g)填充到一个柱反应管中,并将柱反应管与泵相连。然后,将0.006mol的糠醛和0.0072mol的氰乙酸乙酯加入到10ml的量筒中,然后将体积比为25∶1的乙醇/dmf溶剂滴加到量筒中,直到量筒中反应液的体积为10ml。将10ml反应液转移到柱反应管中,并将泵中的流速设置为0.25ml/min,反应液在室温下流反应40min。反应结束后,将生成液进行旋转蒸发操作以获得粗产品,然后经过乙醇重结晶、干燥得到1.04g目标产物2-氰基-3-(呋喃-3-基)丙烯酸乙酯,收率为91%。
与传统方法制备的化合物相比,该路线所得产品纯度更高,熔程更短。该化合物的表征结果如下:m.p.91.6–92.3℃;ft-ir(kbrdisc)cm-1:2222.1,1712.5,1617.0,1209.7,1018.2;1hnmr(500mhz,cdcl3)δ:8.04(s,1h),7.77(d,j=1.6hz,1h),7.41(d,j=3.7hz,1h),6.68(dd,j=3.7,1.7hz,1h),4.38(q,j=7.1hz,2h),1.40(t,j=7.1hz,3h).
上述实施例以0.006mol的苯甲醛和0.0072mol的丙二腈为模型反应进行了最优催化剂和最佳溶剂条件的探讨。实验结果表明,在溶剂为乙醇/dmf(体积比:25∶1)、催化剂为1,6-己二胺功能化氯球(ps-dah)、流速为0.25ml/min的条件下,苄烯丙二腈的收率最高(高达97.4%)。在催化剂的循环次数和底物扩展实验中,ps-dah表现出高稳定性和催化活性,循环10次后产率仍然在85%左右,并且所有的底物扩展实验中,合成α,β-不饱和化合物的收率都在86%以上。
传统的化学合成存在液相传质、传热不均和大量有机溶剂浪费等问题,这不仅降低了产品的品质和收率,同时还造成明显的环境污染。近年来,连续流技术由于其高效的传质传热方式以及对溶剂用量需求降低等优点,已经成为一种高效、绿色的合成技术。本发明一种基于异相碱性催化剂的α,β-不饱和化合物的连续流制备方法,将α,β-不饱和化合物的合成与一种易于商品化、产业化的流技术结合起来。相对于传统的α,β-不饱和化合物的合成方法,具有以下优势:1、相对于传统的cdi2、吡啶、羟基磷灰石等,ps-dah不仅具有高催化活性和稳定性,而且可通过简单洗涤、干燥即可回收再使用、产品通过重结晶纯化,后处理简单且对处理过程不需使用大量有机溶剂进行柱色谱分离,环境友好;2、流技术的运用,提高了产品的纯度、降低了生产成本以及易于α,β-不饱和化合物的快速大批量生产,具有非常好的产业化条件。
上述实施例和附图并非限定本发明的方法,任何所属技术领域的普通技术人员对其所做的适当变化或修饰,皆应视为不脱离本发明的专利范畴。
- 还没有人留言评论。精彩留言会获得点赞!