玉米小斑病菌致病力分化的高通量鉴定方法与流程
本发明涉及一种鉴定方法,具体涉及一种玉米小斑病菌致病力分化的高通量鉴定方法。
背景技术:
玉米是我国主要的粮食作物之一,同时又是非常重要的饲料作物,叶部病害的严重发生是限制玉米生产的主要因素,不但给玉米种植业造成很大的经济损失,而且对养殖业也具有较大的影响。
玉米叶部病害主要是指病原细菌或真菌通过定殖、侵染玉米叶部组织,引起玉米的局部乃至全株的叶片发生病害,可导致玉米局部或全株死亡,严重降低玉米产量甚至绝收。玉米小斑病菌是由cochliobolusheterostrophus(c.heterostrophus)侵染引起的,是一种普遍流行的世界性病害,美国70年代曾因该病害病原菌t小种的流行导致了很严重的损失。玉米小斑病菌根据对雄性细胞质不育系(cytoplasmicmalesterileline,cms)有无专化致病性分为四个生理小种:t小种、c小种、和o小种,o小种在正常细胞质自交系上造成的病斑与其在t、c细胞质不育系上的表现差异不明显,无寄主专化性,且近年在我国南方甜玉米产区o小种对感病甜玉米和青贮玉米品种造成较重危害,同时我国引进的美洲品系不断增加,田间调查发现国外品种的小斑病抗性大大低于国内品种,因此需要加强玉米小斑病的预警和基础研究工作。
本课题组从2012年至2016年连续5年进行了全国玉米小斑病菌的致病力分化监测,构建了一套小种分化鉴定技术,即分别从全国各地采集玉米病叶,进行单孢分离纯化,然后再将纯化的单菌株分别回接到玉米叶片上,对病菌侵染形成的病斑大小进行统计来进行致病力的评估,病斑的大小是通过pdquest软件量化来计算,根据量化的结果制定标准,将致病力分为强、弱不同的致病力。但是由该体系来鉴定玉米小斑病菌致病力费时费力,且回接玉米叶片时,玉米的品种和品种的单一性都会影响致病力的判断,另外,病斑的大小是在病菌侵染玉米叶片3天时进行统计,侵染观察时间点比较单一,缺乏对侵染过程进行测定。因此,该体系不能全面分析玉米小斑病菌致病力的分化。急需开发一种能够在遗传水平上更全面的分析小斑病菌致病力分化的高通量鉴定技术。
技术实现要素:
本发明的目的在于克服上述现有技术中缺乏快速简捷的能全面分析小斑病菌致病力的鉴定技术,提供一种更全面的玉米小斑病菌致病力分化的高通量鉴定方法。
本发明的目的是通过以下技术方案来实现的:
本发明涉及一种玉米小斑病菌致病力分化的高通量鉴定方法,所述方法包括如下步骤:
s1、采集并纯化玉米小斑病菌菌株;
s2、对纯化后的菌株分别进行分子标记基因的pcr扩增和电泳检测;
s3、统计该菌株中分子标记基因的出现概率,若全部分子标记基因出现概率≥70%,则其为强致病型菌株;若全部分子标记基因出现概率<70%,则其为弱致病型菌株。
本发明中,步骤s2中,所述分子标记基因为geneid如下的20个标记基因:
e_gw1.10.1683.1、e_gw1.10.1141.1、estext_genewise1.c_10_t80018、
estext_genewise1plus.c_10_t70478、estext_genewise1plus.c_10_t70486、
estext_genewise1plus.c_6_t30211、gm1.4880_g、
fgenesh1_kg.10_#_336_#_2001_2_ccbo_ccbp_ccbu_ccbw_exta_extb、
gw1.10.732.1、fgenesh1_pm.10_#_1222、e_gw1.23.321.1、estext_genewise1.c_5_t10076、
estext_fgenesh1_pm.c_250021、estext_fgenesh1_pm.c_260013、
cochec5_1.fgenesh1_pg.c_scaffold_26000001、cochec5_1.e_gw1.20.139.1、
fgenesh1_kg.8_#_20_#_2001_2_ccbo_ccbp_ccbu_ccbw_exta_extb、
e_gw1.19.223.1、fgenesh1_pm.18_#_2和e_gw1.16.166.1。
本发明中,所述pcr扩增采用的引物包括:针对e_gw1.10.1683.1标记基因的序列如seqidno.1和seqidno.2所示的引物对;针对e_gw1.10.1141.1标记基因的序列如seqidno.3和seqidno.4所示的引物对;针对estext_genewise1.c_10_t80018标记基因的序列如seqidno.5和seqidno.6所示的引物对;针对estext_genewise1plus.c_10_t70478标记基因的序列如seqidno.7和seqidno.8所示的引物对;针对estext_genewise1plus.c_10_t70486标记基因的序列如seqidno.9和seqidno.10所示的引物对;针对estext_genewise1plus.c_6_t30211标记基因的序列如seqidno.11和seqidno.12所示的引物对;针对gm1.4880_g标记基因的序列如seqidno.13和seqidno.14所示的引物对;针对fgenesh1_kg.10_#_336_#_2001_2_ccbo_ccbp_ccbu_ccbw_exta_extb标记基因的序列如seqidno.15和seqidno.16所示的引物对;针对gw1.10.732.1标记基因的序列如seqidno.17和seqidno.18所示的引物对;针对fgenesh1_pm.10_#_1222标记基因的序列如seqidno.19和seqidno.20所示的引物对;针对e_gw1.23.321.1标记基因的序列如seqidno.21和seqidno.22所示的引物对;针对estext_genewise1.c_5_t10076标记基因的序列如seqidno.23和seqidno.24所示的引物对;针对estext_fgenesh1_pm.c_250021标记基因的序列如seqidno.25和seqidno.26所示的引物对;针对estext_fgenesh1_pm.c_260013标记基因的序列如seqidno.27和seqidno.28所示的引物对;针对cochec5_1.fgenesh1_pg.c_scaffold_26000001标记基因的序列如seqidno.29和seqidno.30所示的引物对;针对cochec5_1.e_gw1.20.139.1标记基因的序列如seqidno.31和seqidno.32所示的引物对;针对fgenesh1_kg.8_#_20_#_2001_2_ccbo_ccbp_ccbu_ccbw_exta_extb标记基因的序列如seqidno.33和seqidno.34所示的引物对;针对e_gw1.19.223.1标记基因的序列如seqidno.35和seqidno.36所示的引物对;针对fgenesh1_pm.18_#_2标记基因的序列如seqidno.37和seqidno.38所示的引物对;针对e_gw1.16.166.1标记基因的序列如seqidno.39和seqidno.40所示的引物对。具体见表1。
本发明中,步骤s1中,所述采集为采集玉米小斑病菌侵染过的玉米叶片。
本发明中,步骤s1中,所述纯化为从玉米小斑病菌侵染过的玉米叶片上,利用单胞分离纯化方式分离纯化出玉米小斑病菌。
本发明中,步骤s2中,所述pcr扩增的条件为:94℃,3min后,94℃,30s,60℃,30s,72℃,30s35个循环,再72℃,5min。
本发明中,步骤s2中,所述电泳检测为1%琼脂糖凝胶电泳检测。
与现有技术相比,本发明具有如下有益效果:
1、本发明的高通量鉴定方法能够在遗传分子水平上更全面的评估玉米小斑病菌的强弱致病,不受寄主差异的影响,评估结果可靠性较高;
2、本发明的高通量鉴定方法能够在大样本量上、短时间内对玉米小斑病菌致病力的强弱进行鉴定,省时省力;
3、本发明的高通量鉴定方法具有易优化性,在此基础上进行优化各种操作条件,可使得操作更简单化、广适化。
附图说明
通过阅读参照以下附图对非限制性实施例所作的详细描述,本发明的其它特征、目的和优点将会变得更明显:
图1为dy-gfp,wf-gfp突变株的构建图谱;
图2为dy和wf的形态及侵染过程的观察示意图;其中,a为dy和wf在培养基上的形态,b为dy和wf在玉米叶片上的病斑形态,c为dy和wf分别在不同时间点的侵染状态图(dm:粗壮致密的菌丝);
图3为dy和wf侵染不同时间下的全转录组分析;其中,a为venn图分析dy和wf中mrna,circrna,lncrna的差异表达;b为dy和wf中差异表达的基因进行heatmap分析;c为参与侵染不同阶段显著差异基因,图中co代表孢子阶段,sm代表稀疏松散菌丝,dm代表粗壮致密阶段;
图4为强弱致病菌dy和wf基因组重测序结果分析;其中,a为dy和wf中拷贝数目变异(cnv)的分析;b为dy和wf中碱基插入/缺失突变(indel)的分析;c为dy和wf中单核苷酸变异(snv)的分析;
图5为基因组和转录组中的差异表达基因的交集基因的kegg分析;
图6为对从湖北省采集分离出来的小斑病菌致病情况的鉴定比较,上图采用叶片回接法鉴定;下图采用本发明的高通量的鉴定方法;
图7为对从河南省新乡市采集分离出来的小斑病菌致病情况的鉴定比较,上图采用叶片回接法鉴定;下图采用本发明的高通量的鉴定方法;
图8为对从河南省农科院采集分离出来的小斑病菌致病情况的鉴定比较,上图采用叶片回接法鉴定;下图采用本发明的高通量的鉴定方法;
图9为对从河南省漯河市采集分离出来的小斑病菌致病情况的鉴定比较,上图采用叶片回接法鉴定;下图采用本发明的高通量的鉴定方法;
图10为对从河北省衡水市采集分离出来的小斑病菌致病情况的鉴定比较,上图采用叶片回接法鉴定;下图采用本发明的高通量的鉴定方法;
图11为对从河北省衡水市采集分离出来的小斑病菌致病情况的鉴定比较,上图采用叶片回接法鉴定;下图采用本发明的高通量的鉴定方法;
图12为对从河北省唐山市采集分离出来的小斑病菌致病情况的鉴定比较,上图采用叶片回接法鉴定;下图采用本发明的高通量的鉴定方法;
图13为对从河北省唐山市采集分离出来的小斑病菌致病情况的鉴定比较,上图采用叶片回接法鉴定;下图采用本发明的高通量的鉴定方法;
图14为对从河北省衡水市采集分离出来的小斑病菌致病情况的鉴定比较,上图采用叶片回接法鉴定;下图采用本发明的高通量的鉴定方法;
图15为对从河南省新乡市采集分离出来的小斑病菌致病情况的鉴定比较,上图采用叶片回接法鉴定;下图采用本发明的高通量的鉴定方法。
具体实施方式
下面结合实施例对本发明进行详细说明。以下实施例将有助于本领域的技术人员进一步理解本发明,但不以任何形式限制本发明。应当指出的是,对本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干调整和改进。这些都属于本发明的保护范围。
实施例1
本实施例涉及一种玉米小斑病菌致病力分化的高通量鉴定方法的构建,具体如下:
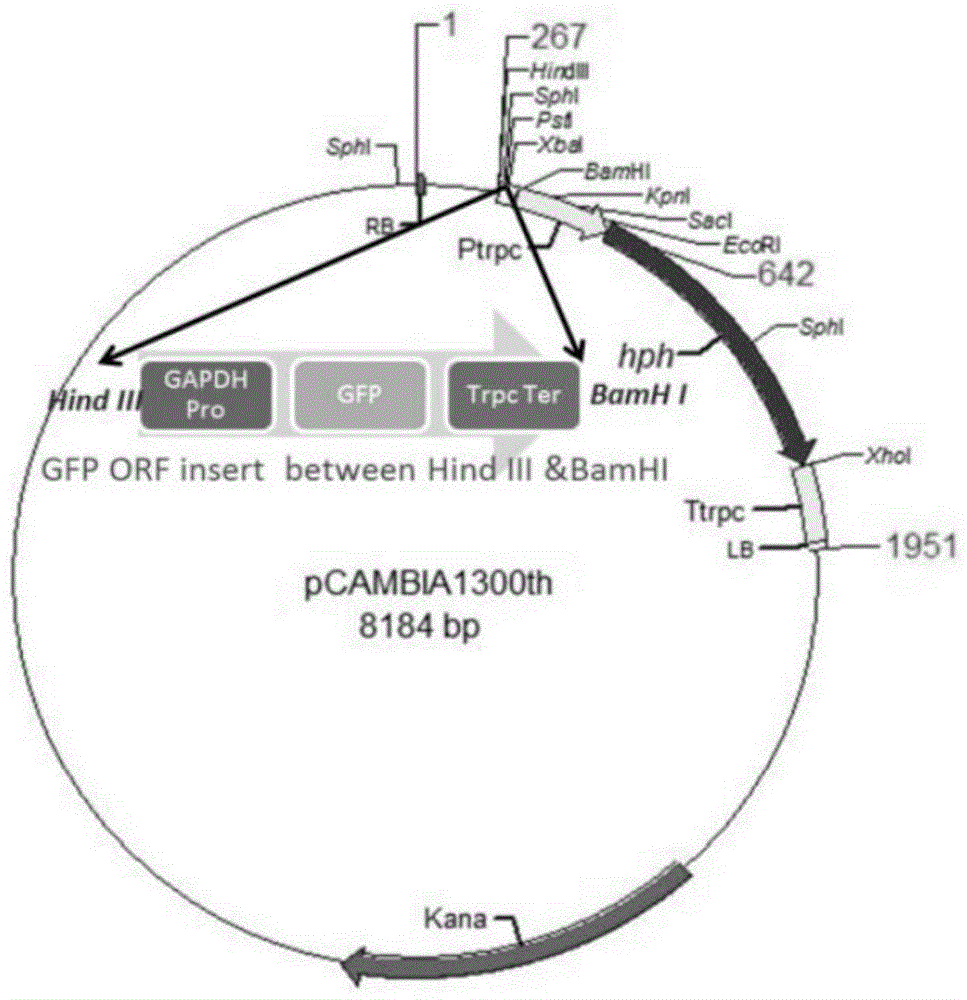
发明人从田间分离到的菌株中选取了致病力差异较显著的玉米小斑病菌不同的致病力类型:dy,强致病菌株;wf,弱致病菌株(其中,dy是从浙江省东阳市田间的玉米病叶上经过单孢分离纯化得到的小斑病菌,再回接到玉米叶片后通过统计病斑大小的方法鉴定该菌株为强致病菌株;wf是从山东省潍坊市田间的玉米病叶上采集并单孢分离纯化得到的小斑菌株,再回接到玉米叶片后观察病斑的大小的方法,鉴定到该菌株为弱致病菌株)。构建了dy-gfp,wf-gfp突变株(通过农杆菌介导的转化方法(agrobacteriumtumefaciens-mediatedtransformation,atmt)进行构建gfp突变株,骨架质粒pcambia1300th,gfp荧光蛋白表达框启动子采用三磷酸甘油醛脱氢酶(gapdh)强启动子,终止子采用trpcterminator;构建图谱如图1所示。
对其侵染过程进行了观察与分析,发现孢子萌发后,强致病菌株dy的芽管与气生菌丝生长速度均快于弱致病菌株wf。侵染玉米9小时后,dy菌株松散无序的菌丝尖端开始变得粗壮,侵染开始发生转变,是基因调动最剧烈的阶段,致病力分化菌株中基因调动的策略也会有很大的不同(图2)。
因此,本发明选取孢子阶段、侵染准备阶段和菌斑扩展阶段这三个阶段进行全转录组分析,包括mrna,lncrna和circrna。对强弱致病菌株中的表达有差异的基因进行聚类分析,另外借助于7个致病因子数据库:p450、转运蛋白tcdb、转录因子ftfd、病原寄主互作、小分泌蛋白ssp、非核糖体多肽合成酶nrps和pks,进行差异基因功能预测,然后对各个侵染阶段的差异基因功能进行分析,发现在强致病力菌株dy中孢子启动阶段显著升高的基因有gtp酶活化子因子、磷酸化酶、激酶和几丁质合酶等,在侵染准备阶段显著差异的基因有丝氨酸、羧肽酶、氧化还原酶和双加氧酶等,在菌斑扩展阶段差异基因与侵染准备阶段差不多,另外水解酶也呈现显著差异(图3)。可见,在全转录组上的分析结果显示致病力的分化在小斑侵染玉米过程早期就出现了,中期和后期都分别表现出很大的差异。
接下来又对强弱致病菌进行基因组重测序,对强弱致病菌株基因组上的差异进行分析。分析结果显示(图4),由拷贝数目变异导致的基因变异数是最多的,与强致病菌株dy相比较,弱致病菌株wf中缺失的基因较多,因此拷贝数目的变异可能是导致致病力差异的主效因素。
将基因组上的差异变化和转录组中的差异变化基因,进行交集筛选,然后对这些基因可能参与的代谢途径进行kegg分析,发现这些差异基因主要分布在内质网上蛋白质的加工过程,氧化磷酸化,和脂肪酸降解的代谢途径中(图5),然后将这三条途径中所涉及到的基因进行筛选,选出差异最显著的,作为最有可能全面反映o小种致病力分化的分子标记物。
然后筛选出20个与致病力相关性较高的基因作为分子标记物,选用2017年叶片回接法分离鉴定出的强弱致病力不同的菌株进行这20个基因的出现频率的统计分析(2017年的菌株是发明人所在课题组通过从全国各地采集分离的菌株,用叶片回接法评估的菌株的致病力,然后分别用20%甘油保存于-80℃冰箱。2018年活化后提取dna,进行标记基因的统计的。此外,本发明人也在2018年全国各地收集到的菌株中随机选取几株菌进行叶片回接法和标记基因统计法进行比较,结果相一致,说明标记基因选取具备可靠性)。通过20个标记基因(20个标记基因见表1)出现概率进行统计分析,制定了鉴定标准:20个标记基因在纯化后的小斑病菌菌株中出现的概率若≥70%,则将其划分为强致病型菌株;出现的概率若<70%,则将其划分为弱致病型菌株。
至此,本实施例的玉米小斑病菌致病力分化的高通量鉴定方法,具体包括如下步骤
s1、采集并纯化玉米小斑病菌菌株;
s2、对纯化后的菌株分别进行所述分子标记基因的pcr扩增和电泳检测。
s3、统计该菌株中分子标记基因的出现概率,若全部分子标记基因出现概率≥70%,则其为强致病型菌株;若分子标记基因出现概率<70%,则其为弱致病型菌株。
表1本发明中所筛选的20个标记基因及鉴定用的引物信息
参考基因组下载链接为:http://genome.jgi.doe.gov/cochec5_3/cochec5_3.home.html
实施例2
本实施例涉及本发明的玉米小斑病菌致病力分化的高通量鉴定方法的效果验证。
按照强弱致病株鉴定标准,对2018年采集并纯化到的玉米小斑病菌(具体是从全国各地采集病叶,将病叶用次氯酸钠进行表面消毒后接在pda培养基中培养2天,然后挑取菌丝尖端到新的pda培养基中培养5-7天,待纯化的小斑菌产孢子后,进行单孢分离纯化,即得到纯化的小斑病菌)随机挑选10株进行验证。分别用传统的叶片回接法和本发明的高通量检测方法鉴定分离纯化的菌株的致病力。
玉米品种用四种不同小斑病菌抗性的品种:b73感病品种、mo17中抗性品种、x178和黄早四抗性较mo17低。
图6为对从湖北省采集分离出来的小斑病菌致病情况的鉴定比较,上图采用叶片回接法鉴定,分别将纯化后的小斑病菌回接到不同抗性的玉米品种上(b73(感病品种),mo17(中抗品种),x178(中抗品种),黄早四(中抗品种)),侵染3天后进行病斑大小的统计,评估该菌株的侵染致病力为弱致病力;下图采用本发明的高通量的鉴定方法,20个分子标记基因在该菌株中出现的额概率为20%,远远小于70%,根据高通量鉴定方法的判定标准,该菌株为弱致病菌株;与传统的叶片回接方法得到的结果一致。
图7为对从河南省新乡市采集分离出来的小斑病菌致病情况的鉴定比较,上图采用叶片回接法鉴定(同图1的描述)侵染3天后进行病斑大小的统计,评估该菌株的侵染致病力为弱致病力。下图采用本发明的高通量的鉴定方法,20个分子标记基因在该菌株中出现的额概率为40%,小于70%,根据高通量鉴定方法的判定标准,该菌株为弱致病菌株。与传统的叶片回接方法得到的结果一致。
图8为对从河南省农科院采集分离出来的小斑病菌致病情况的鉴定比较,上图采用叶片回接法鉴定(同图1的描述)侵染3天后进行病斑大小的统计,评估该菌株的侵染致病力为强致病力。下图采用本发明的高通量的鉴定方法,20个分子标记基因在该菌株中出现的额概率为80%,大于70%,根据高通量鉴定方法的判定标准,该菌株为强致病菌株。与传统的叶片回接方法得到的结果一致。
图9为对从河南省漯河市采集分离出来的小斑病菌致病情况的鉴定比较,上图采用叶片回接法鉴定(同图1的描述)侵染3天后进行病斑大小的统计,评估该菌株的侵染致病力为强致病力。下图采用本发明的高通量的鉴定方法,20个分子标记基因在该菌株中出现的额概率为70%,等于70%,根据高通量鉴定方法的判定标准,该菌株为强致病菌株。与传统的叶片回接方法得到的结果一致。
图10为对从河北省衡水市采集分离出来的小斑病菌致病情况的鉴定比较,上图采用叶片回接法鉴定(同图1的描述)侵染3天后进行病斑大小的统计,评估该菌株的侵染致病力为强致病力。下图采用本发明的高通量的鉴定方法,20个分子标记基因在该菌株中出现的额概率为90%,大于70%,根据高通量鉴定方法的判定标准,该菌株为强致病菌株。与传统的叶片回接方法得到的结果一致。
图11为对从河北省衡水市采集分离出来的小斑病菌致病情况的鉴定比较,上图采用叶片回接法鉴定(同图1的描述)侵染3天后进行病斑大小的统计,评估该菌株的侵染致病力为弱致病力。下图采用本发明的高通量的鉴定方法,20个分子标记基因在该菌株中出现的额概率为25%,小于70%,根据高通量鉴定方法的判定标准,该菌株为弱致病菌株。与传统的叶片回接方法得到的结果一致。
图12为对从河北省唐山市采集分离出来的小斑病菌致病情况的鉴定比较,上图采用叶片回接法鉴定(同图1的描述)侵染3天后进行病斑大小的统计,评估该菌株的侵染致病力为弱致病力。下图采用本发明的高通量的鉴定方法,20个分子标记基因在该菌株中出现的额概率为55%,小于70%,根据高通量鉴定方法的判定标准,该菌株为弱致病菌株。与传统的叶片回接方法得到的结果一致。
图13为对从河北省唐山市采集分离出来的小斑病菌致病情况的鉴定比较,上图采用叶片回接法鉴定(同图1的描述)侵染3天后进行病斑大小的统计,评估该菌株的侵染致病力为强致病力。下图采用本发明的高通量的鉴定方法,20个分子标记基因在该菌株中出现的额概率为80%,大于70%,根据高通量鉴定方法的判定标准,该菌株为强致病菌株。与传统的叶片回接方法得到的结果一致。
图14为对从河北省衡水市采集分离出来的小斑病菌致病情况的鉴定比较,上图采用叶片回接法鉴定(同图1的描述)侵染3天后进行病斑大小的统计,评估该菌株的侵染致病力为强致病力。下图采用本发明的高通量的鉴定方法,20个分子标记基因在该菌株中出现的额概率为70%,等于70%,根据高通量鉴定方法的判定标准,该菌株为强致病菌株。与传统的叶片回接方法得到的结果一致。
图15为对从河南省新乡市采集分离出来的小斑病菌致病情况的鉴定比较,上图采用叶片回接法鉴定(同图1的描述)侵染3天后进行病斑大小的统计,评估该菌株的侵染致病力为弱致病力。下图采用本发明的高通量的鉴定方法,20个分子标记基因在该菌株中出现的额概率为55%,小于70%,根据高通量鉴定方法的判定标准,该菌株为弱致病菌株。与传统的叶片回接方法得到的结果一致。
此外,尽管通过上述两种方法的结果的对比,得到的结论是叶片回接法与本发明的高通量方法鉴定的小斑病菌致病力分化的结果是一致的,但是叶片回接法操作复杂,费时费力,且受鉴别寄主的影响非常大。而高通量较叶片回接法大大缩短了鉴定时间,且鉴定操作简单,且不会受鉴别寄主的影响,能够全面反应致病力的分化。
实施例3
本实施例涉及本发明的玉米小斑病菌致病力分化的高通量鉴定方法的应用案例:
河南省
河南省共鉴定到36株小斑病菌,用该高通量鉴定技术鉴定到强致病株有12株,弱致病株有24株,分别占33.3%和66.7%。
实施例4
本实施例涉及本发明的玉米小斑病菌致病力分化的高通量鉴定方法的应用案例:
河北省
河北省共鉴定到24株小斑病菌,用该高通量鉴定技术鉴定到强致病株有10株,弱致病株有14株,分别占41.7%和58.3%。
以上对本发明的具体实施例进行了描述。需要理解的是,本发明并不局限于上述特定实施方式,本领域技术人员可以在权利要求的范围内做出各种变形或修改,这并不影响本发明的实质内容。
序列表
<110>上海交通大学
<120>玉米小斑病菌致病力分化的高通量鉴定方法
<130>dag38185
<160>40
<170>siposequencelisting1.0
<210>1
<211>18
<212>dna
<213>人工序列(artificialsequence)
<400>1
ctcttccgtaggcgtaac18
<210>2
<211>19
<212>dna
<213>人工序列(artificialsequence)
<400>2
aatgtagcagtggcgtatt19
<210>3
<211>19
<212>dna
<213>人工序列(artificialsequence)
<400>3
actcgctcatctacttcct19
<210>4
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>4
gctacttgttgctcttcttg20
<210>5
<211>18
<212>dna
<213>人工序列(artificialsequence)
<400>5
atgaacctgcgacaagac18
<210>6
<211>19
<212>dna
<213>人工序列(artificialsequence)
<400>6
atgatgccaattccaccaa19
<210>7
<211>21
<212>dna
<213>人工序列(artificialsequence)
<400>7
cattgttgtccgtatccttag21
<210>8
<211>18
<212>dna
<213>人工序列(artificialsequence)
<400>8
ctcgtctggcttgttcat18
<210>9
<211>21
<212>dna
<213>人工序列(artificialsequence)
<400>9
gacatccataatccaccagta21
<210>10
<211>18
<212>dna
<213>人工序列(artificialsequence)
<400>10
cctcctcaccacgaagaa18
<210>11
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>11
ccgatgaggttgttacagta20
<210>12
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>12
ccgtgagacaggtaatgaat20
<210>13
<211>18
<212>dna
<213>人工序列(artificialsequence)
<400>13
gctatggtgccttcaact18
<210>14
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>14
agcgtgtatccagtattctt20
<210>15
<211>18
<212>dna
<213>人工序列(artificialsequence)
<400>15
tcgtcgttgaagaaggag18
<210>16
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>16
cggaccagttacttatacac20
<210>17
<211>18
<212>dna
<213>人工序列(artificialsequence)
<400>17
acgacaacgctcttatgc18
<210>18
<211>21
<212>dna
<213>人工序列(artificialsequence)
<400>18
tctaatccgctaatgacttcc21
<210>19
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>19
agaacctgattgatgcttgt20
<210>20
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>20
accacctaagtagacagagt20
<210>21
<211>18
<212>dna
<213>人工序列(artificialsequence)
<400>21
atccagttccaggtccaa18
<210>22
<211>18
<212>dna
<213>人工序列(artificialsequence)
<400>22
agtgcttctcaggtccat18
<210>23
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>23
tatcacagagccaagtatcc20
<210>24
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>24
gccaacgaatatcaggtatg20
<210>25
<211>18
<212>dna
<213>人工序列(artificialsequence)
<400>25
agtgctggcgatgtaatg18
<210>26
<211>19
<212>dna
<213>人工序列(artificialsequence)
<400>26
gttgctcctgtatggtgta19
<210>27
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>27
ggagcggtatcaatcctatt20
<210>28
<211>19
<212>dna
<213>人工序列(artificialsequence)
<400>28
aacggatgctctaccaatt19
<210>29
<211>21
<212>dna
<213>人工序列(artificialsequence)
<400>29
cggaagttattagcggattag21
<210>30
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>30
taggttcgtaggagttgttc20
<210>31
<211>18
<212>dna
<213>人工序列(artificialsequence)
<400>31
aacactgagcgaagaaca18
<210>32
<211>18
<212>dna
<213>人工序列(artificialsequence)
<400>32
ctgaagaggtagcgagag18
<210>33
<211>18
<212>dna
<213>人工序列(artificialsequence)
<400>33
catcgtcattccgttggt18
<210>34
<211>19
<212>dna
<213>人工序列(artificialsequence)
<400>34
ctcagcctcttgttgttca19
<210>35
<211>18
<212>dna
<213>人工序列(artificialsequence)
<400>35
gcggttgccttgtaagat18
<210>36
<211>18
<212>dna
<213>人工序列(artificialsequence)
<400>36
gaccaatccacagtagcc18
<210>37
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>37
aggatagaaggtctctgtga20
<210>38
<211>18
<212>dna
<213>人工序列(artificialsequence)
<400>38
gctaggtctgcctctgta18
<210>39
<211>18
<212>dna
<213>人工序列(artificialsequence)
<400>39
agaactggagacgcaaga18
<210>40
<211>22
<212>dna
<213>人工序列(artificialsequence)
<400>40
ggatctgaactgtattgattgg22
- 还没有人留言评论。精彩留言会获得点赞!