一种一步光催化木质素解聚及胺化合成含氮芳香化合物的方法与流程
本发明属于木质素应用技术领域,具体涉及一种一步光催化木质素解聚及胺化合成含氮芳香化合物的方法。
背景技术:
石油是一种不可再生资源,寻求可持续的石油替代品是顺应当代社会可持续发展的需要。木质纤维素是地球上最丰富的含碳生物质,它主要由纤维素、半纤维素以及木质素组成。其中,木质素的储量仅次于纤维素,是第二多可再生生物质资源,也是自然界唯一的含芳基的非化石基资源。但是,木质素至今也没有得到大量而广泛地应用,其大多作为造纸废水被排放,或者焚烧,这不仅造成严重的环境污染,更是白白浪费掉大自然恩赐给人类的一大资源。
木质素是由三种苯丙烷结构单体以c-o键或c-c键连接而成的生物大分子,其通过一定的共价键和氢键与纤维素和半纤维素交联在一起组成了三维空间交联结构,使植物细胞壁具有足够的强度以保护植物细胞。木质素的结构十分复杂,是由苯基丙烷类结构单元(紫丁香基丙烷单元s、愈创木基丙烷单元g、对羟基苯基丙烷单元h)通过碳-碳键和醚键连接而成的具有三维网状结构的高分子聚合物。木质素的复杂结构、难提取分离特性和稳定的化学键使得其降解应用非常困难。目前,由于c-o键的低能垒,木质素的降解主要集中在c-o键的断裂上,已经开发出多种有效的c-o键断裂的方法,例如,氢化、氧化、歧化、酸解、碱解等。而c-c键的惰性和较高的能垒,其断裂存在着巨大的挑战。目前c-c键的裂解主要集中在热诱导体系(钒、铜、铁、铱、铑、钌等金属催化)和光诱导体系(钒、铱等金属催化和一些复合纳米材料)上。
目前,木质素c-c键的裂解存在以下几个问题:1)反应条件苛刻,需要高温或高压;2)反应步骤繁琐,一般需要经历多步反应;3)断键选择性差,通常伴随着c-o键的裂解;4)产物选择性差,会产生多种产物。因此,实现温和条件下高效、高选择性地断裂木质素c-c键的催化体系还有待开发。
技术实现要素:
本发明要解决的技术问题在于,克服背景技术的不足,提供一种步骤简单、反应条件温和的高效、高选择性的一步光催化木质素解聚及胺化合成含氮芳香化合物的方法。
一种一步光催化木质素解聚及胺化合成含氮芳香化合物的方法,其特征在于:以木质素或其模型化合物作为反应底物,将木质素或其模型化合物溶于溶剂,在光源激发的条件下,在光催化剂催化下,在助剂及氮源的辅助下,进行c-c键的选择性裂解及胺化,从而合成含氮芳香化合物;木质素或其模型化合物、光催化剂、助剂、氮源的摩尔比100:(1~20):(1~100):100,反应温度为室温,反应时间为6~24小时。
所述光源为蓝光(λ=407nm~505nm)led灯、紫光(λ=380nm~470nm)led灯、紫外(λ=200nm~380nm)led灯等;
所述光催化剂为三氯化铈、三溴化铈、二正四丁基六氯化铈[(nbu4n)2cecl6]、三氟甲磺酸铈等;
所述助剂为四丁基氯化铵、四丁基氟化铵、四丁基溴化铵、四丁基碘化铵、四丁基氯化磷、四丁基氟化磷、四丁基溴化磷、四丁基碘化磷等;
所述氮源为偶氮二甲酸二乙酯、偶氮二甲酸二异丙酯、偶氮二甲酸二异叔丁酯等;
所述的溶剂的用量为使反应底物的浓度为0.1~5mol/l,所述的有机溶剂为二氯甲烷、乙醚、四氢呋喃、甲苯、乙腈、丙酮、吡啶、n,n-二甲基甲酰胺等。
所述的木质素或其模型化合物为如下具有α-oh的β-o-4键、β-5键和β-1键结构单元的木质素二聚体、三聚体、多聚体、天然松木木质素、桦木木质素、山毛榉木质素、秸秆木质素等各种类型的木质素。
本发明利用铈化合物为光催化剂,在光、助剂及氮源的协同下,通过配体到金属的电荷转移反应活化木质素α-oh得到烷氧自由基,烷氧自由基弱化其邻位的c-c键,同时在偶氮类化合物的作用下实现温和条件下木质素c-c键的裂解及胺化。综上,本发明有以下有益效果:
1、本发明的催化体系操作方便、反应条件温和、转化率高(可达100%)。
2、本发明的催化体系催化剂使用量少(底物与催化剂的摩尔比可以达到500:1),底物适用性广泛。
3、本发明的催化体系选择性裂解c-c键而保留c-o键,经c-c键裂解后,只得到两种产物,原子效率100%。
4、本发明的催化体系可以在温和条件下一步反应即可实现c-c键的裂解及胺化。
5、本发明的催化体系可以实现天然松木木质素、山毛榉木质素、桦木木质素、秸秆等木质素的c-c键裂解,有利于木质素降解的大规模工业化生产和应用。
6、本发明的催化体系可循环连续加料十次。
7、本发明的催化体系具有光可控性,即光照下反应稳步进行,而无光照下反应暂停。
8、本发明的催化体系得到的肼类含氮芳香化合物经两步反应可以环化,大大拓展了产物的应用。
附图说明
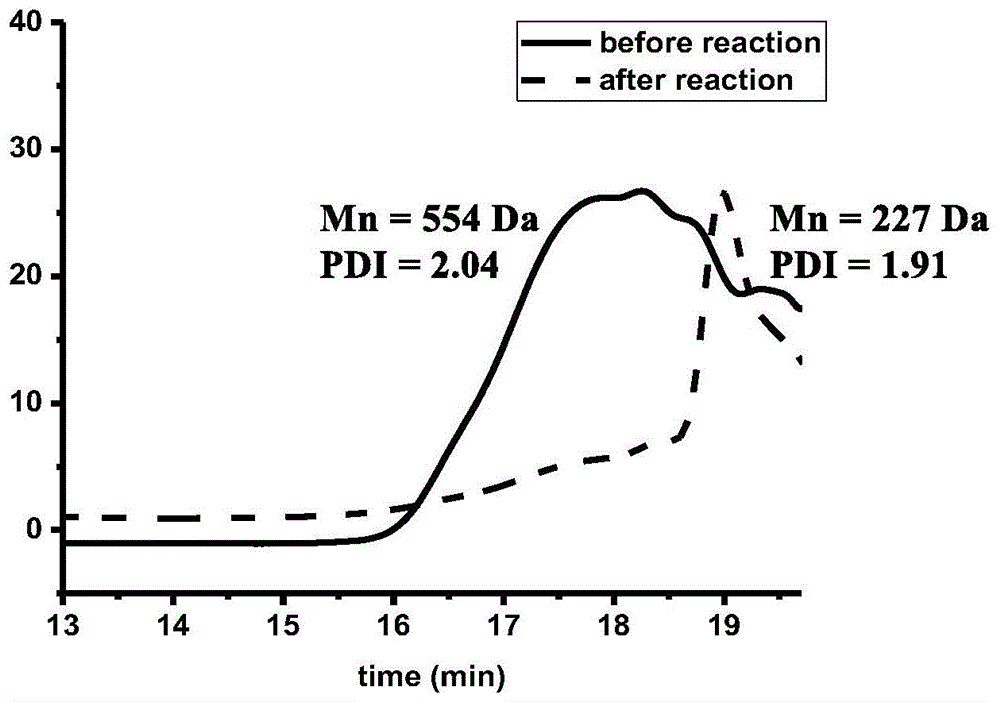
图1:松木木质素反应前后的gpc图;将反应耐压管密闭置于蓝色led灯下,搅拌12小时,松木木质素的分子量从554g/mol降至227g/mol。
图2:桦木木质素反应前后的gpc图;将反应耐压管密闭置于蓝色led灯下,搅拌12小时,桦木木质素的分子量从618g/mol降至246g/mol。
图3:木质素二聚体5的连续加料反应的转化率及产率情况图:第十次加料反应结束后,依然能够得到80%的苯甲醛及79%的肼1。
具体实施方式
通过以下实施例可以进一步说明本发明,实施例是为了说明本发明而不是限制本发明,本发明的保护范围不限制于此。
实施例1:木质素二聚体1的选择性c-c键的裂解及胺化
反应加料在手套箱中进行,称取木质素二聚体1(0.1mmol,24.4mg)、光催化剂三氯化铈(1mol%)、助剂四丁基氯化铵(5mol%)、偶氮二甲酸二叔丁酯(0.1mmol)和0.5ml乙腈于10毫升反应耐压管中。将反应耐压管密闭置于蓝灯420nm下,搅拌12小时待木质素二聚体1完全转化后,经硅胶柱层析(乙酸乙酯:石油醚=1:5,体积比)分离后得到产物对甲氧基苯甲醛(12.4mg,产率91%)和1-(苯氧基甲基)-1,2-二羧酸二叔丁酯基-肼(肼1)(30.8mg,产率91%)。
核磁共振数据:
对甲氧基苯甲醛:1hnmr(500mhz,chloroform-d)δ9.87(s,1h),7.81–7.75(m,2h),7.01–6.95(m,2h),3.80(s,3h).13cnmr(126mhz,cdcl3)δ190.8,166.6,132.1,130.5,114.7,55.3。
肼1:1hnmr(500mhz,chloroform-d)δ7.27(t,j=7.0hz,2h),7.01–6.97(m,3h),6.65–6.33(m,1h),5.38(s,2h),1.47–1.43(m,18h).13cnmr(126mhz,cdcl3)δ155.3,137.3,128.6,127.6,81.4,54.5,53.0,28.3,28.3。
实施例2木质素二聚体2的选择性c-c键的裂解及胺化
反应加料在手套箱中进行,称取木质素二聚体2(0.1mmol,27.4mg)、光催化剂二正四丁基六氯化铈[(nbu4n)2cecl6](1mol%)、助剂四丁基氯化磷(5mol%)、氮源偶氮二甲酸二异丙酯(0.1mmol)和1.0ml二氯甲烷于10毫升耐压管中。将反应耐压管密闭置于紫外365nm下,搅拌18小时待木质素二聚体2完全转化后,经硅胶柱层析(乙酸乙酯:石油醚=1:5,体积比)分离后得到产物对甲氧基苯甲醛(12.5mg,产率92%)和1-(1-苯氧基乙醇基)-1,2-二羧酸二异丙酯基-肼(肼2)(20.4mg,产率81%)。
核磁共振数据:
对甲氧基苯甲醛:如实施例1。
肼2:1hnmr(500mhz,chloroform-d)δ7.34–7.26(m,2h),7.05–6.99(m,2h),6.91(tt,j=7.5,2.1hz,1h),4.98(t,j=7.0hz,1h),4.27(ddd,j=12.5,7.0,5.5hz,1h),4.07(ddd,j=12.5,7.1,5.5hz,1h),3.39(hept,j=6.8hz,1h),3.16(hept,j=6.8hz,1h),2.96(t,j=5.5hz,1h),2.21(s,1h),1.32(ddd,j=39.0,25.0,6.8hz,12h).13cnmr(126mhz,chloroform-d)δ156.5,129.8,122.2,117.2,98.4,64.3,52.5,51.8,22.1,21.6。
实施例3木质素二聚体3的选择性c-c键的裂解及胺化
反应加料在手套箱中进行,称取木质素二聚体3(0.1mmol,33.4mg)、光催化剂三氟甲磺酸铈(5mol%)、助剂四丁基氯化铵(5mol%)、氮源偶氮二甲酸二叔丁酯(1mmol)和2.0ml吡啶于10毫升耐压管中。将反应耐压管密闭置于紫灯380nm下,搅拌24小时待木质素二聚体3完全转化后,经硅胶柱层析(乙酸乙酯:石油醚=1:5,体积比)分离后得到产物3,4-二甲氧基苯甲醛(13.4mg,产率81%)和1-[1-(2-甲氧基)苯氧基乙醇基]-1,2-二羧酸二异丙酯基-肼(肼3)(28.3mg,产率71%)。
核磁共振数据:
3,4-三甲氧基苯甲醛:1hnmr(500mhz,chloroform-d)δ9.85(d,j=1.2hz,1h),7.52–7.43(m,2h),7.03(d,j=7.5hz,1h),3.89(d,j=10.1hz,6h).13cnmr(126mhz,chloroform-d)δ191.3,154.3,149.6,130.1,126.4,110.1,110.0,56.0,56.0。
肼3:1hnmr(500mhz,chloroform-d)δ7.12(t,j=7.5hz,1h),7.06(d,j=7.5hz,1h),6.95–6.92(m,2h),6.73–6.46(m,1h),6.20–5.91(m,1h),4.29–4.06(m,1h),3.95(td,j=12.0,4.0hz,1h),3.88(s,3h),3.84–3.79(m,1h),1.54(s,9h),1.39(s,3h),1.16(s,7h).13cnmr(126mhz,cdcl3)δ157.9,154.5,152.3,145.2,125.3,122.6,121.5,112.4,89.4,82.5,82.5,60.3,56.0,28.1,27.6。
实施例4木质素二聚体4的选择性c-c键的裂解及胺化
改变底物为上式中的4(0.1mmol,19.8mg),其他条件如实施例1,得到苯甲醛(9.1mg,产率86%)和1-苄基-1,2-二羧酸二叔丁酯基-肼(肼4)(28.3mg,产率88%)。
核磁共振数据:
苯甲醛:1hnmr(500mhz,chloroform-d)δ7.88(dd,j=6.7,2.9hz,1h),7.53(qd,j=4.0,1.7hz,2h).13cnmr(125mhz,chloroform-d)δ192.9,137.6,134.7,129.9,129.6。
肼4:1hnmr(500mhz,chloroform-d)δ7.34–7.26(m,5h),6.00–6.25(m,1h),4.64(s,2h),1.48(s,9h),1.44(s,9h).13cnmr(126mhz,cdcl3)δ155.2,137.3,128.6,127.6,81.4,54.5,53.0,28.3,28.3。
实施例5木质素三聚体的选择性c-c键的裂解及胺化
改变底物为上式中的三聚体(0.1mmol,38.0mg),其他条件如实施例1,得到对甲氧基苯甲醛(11.0mg,产率81%)、1-(4-醛基-苯氧基甲基)-1,2-二羧酸二叔丁酯基-肼(肼5)(28.9mg,产率79%)和肼1(26.4mg,产率78%)。
核磁共振数据:
对甲氧基苯甲醛:如实施例1
肼5:1hnmr(500mhz,chloroform-d)δ9.82(s,1h),7.76(d,j=8.0hz,2h),7.05(s,2h),6.46(s,1h),6.17(s,1h),5.40(s,2h),1.41(s,9h),1.40(s,9h).13cnmr(126mhz,cdcl3)δ190.9,161.9,155.9,154.8,132.1,130.7,116.1,115.8,83.0,82.1,81.6,78.9,28.3,28.2。
肼1:如实施例1
实施例6松木木质素的提取、解聚及胺化
将10.0g松木木屑、40ml二氧六环和1.7mlhcl(37wt%)加入圆底烧瓶中,置于85℃的油浴中回流3小时。将体系冷却至室温后,加入3.36gnahco3并将溶液再搅拌30分钟。然后过滤反应混合物并用丙酮洗涤。然后将所得溶液在40℃下减压浓缩。用二氯甲烷(2×20ml)萃取混合物。将有机层用无水硫酸钠干燥并在减压下浓缩至少量液体。为了沉淀木质素,将浓缩液滴加到500ml正在搅拌的己烷中。过滤溶液以回收沉淀物。然后将木质素粉末用己烷(50ml)和乙醚(50ml)洗涤5分钟,同时进行超声处理。然后将回收的木质素在室温下干燥,沉淀粉末的最终质量为1.35g桦木木质素。
在手套箱中,称取松木木质素(20mg)、光催化剂三氯化铈(1mol%)、助剂四丁基氯化铵(5mol%)、氮源偶氮二甲酸二叔丁酯(0.1mmol)和0.5ml乙腈于10毫升耐压管中。将反应耐压管密闭置于蓝色led灯下,搅拌12小时。松木木质素的分子量从554g/mol降至227g/mol(附图1)。经酸解后释放出香草醛,由气相色谱测试后得到产率为1.45wt%的香草醛。
核磁共振数据:
香草醛:1hnmr(500mhz,chloroform-d)δ9.85(d,j=1.2hz,1h),7.32–7.25(m,2h),6.92(d,j=7.3hz,1h),6.10(s,1h),3.84(s,3h).13cnmr(126mhz,cdcl3)δ191.3,152.6,148.0,130.0,127.7,115.1,109.8,56.3。
实施例7桦木木质素的提取、解聚及胺化
桦木木质素的提取步骤参照实施例6。
改变底物为桦木木质素,其他条件如实施例5。桦木木质素的分子量从618g/mol降至246g/mol(附图2)。经酸解后释放出香草醛和丁香醛,由气相色谱测试后得到产率为0.72wt%的香草醛和0.81wt%丁香醛。
核磁共振数据:
香草醛:如实施例6
丁香醛:1hnmr(500mhz,chloroform-d)δ6.98(d,j=1.1hz,2h),5.93(s,1h),3.90(s,6h).13cnmr(126mhz,cdcl3)δ191.8,147.6,138.8,130.4,106.8,56.5。
实施例8连续加料反应
反应加料在手套箱中进行,称取木质素二聚体5(0.1mmol,21.4mg)、光催化剂三氯化铈(1mol%)、助剂四丁基氯化铵(5mol%)、偶氮二甲酸二叔丁酯(0.1mmol)和0.5ml乙腈于10毫升反应耐压管中。将反应耐压管密闭置于蓝灯420nm下,搅拌12小时待木质素二聚体5完全转化后,继续向反应也中加入木质素二聚体5(0.1mmol,21.4mg)和偶氮二甲酸二叔丁酯(0.1mmol),每次加料,反应结束后,转化率及产物的产率情况如图3所示。待第十次加料反应结束后,经硅胶柱层析(乙酸乙酯:石油醚=1:5,体积比)分离后得到产物苯甲醛(84.8mg,产率80%)和1-(苯氧基甲基)-1,2-二羧酸二叔丁酯基-肼(肼1)(267.0mg,产率79%)。
核磁共振数据:
苯甲醛:如实施例4
肼1:如实施例1
实施例9产物肼2的应用:环化
向肼2(0.2mmol,1.0eq.)的二氯甲烷(2ml)溶液中加入4-二甲氨基吡啶(0.02mmol,10mol%)、三乙胺(0.6mmol,3.0eq.)和甲酰氯(0.3mmol,1.5eq.)。将反应混合物在室温下搅拌30分钟,用饱和氯化铵水溶液(2ml)淬灭,然后用二氯甲烷(3×20ml)萃取,用饱和碳酸氢钠水溶液和饱和食盐水洗涤。将有机层合并,并用无水硫酸镁干燥后过滤,滤液真空浓缩。将粗混合物溶于乙腈(3ml)溶剂中,加入碳酸铯(4.0eq.)后,将得到的混合物在室温下搅拌5小时。然后用饱和氯化铵水溶液(1ml)淬灭反应,用乙酸乙酯(3×20ml)萃取,有机相依次用水和饱和食盐水洗涤。将有机层用无水硫酸镁干燥,过滤后并真空浓缩。通过柱层析色谱法(展开剂是乙酸乙酯:己烷=1:10)分离混合产物,得到1,2-二氮杂环丁烷(7.7mg,产率24%)和1,3,4-恶二嗪(19.3mg,产率60%)。
核磁共振数据:
1,2-二氮杂环丁烷:1hnmr(500mhz,chloroform-d)δ7.30(t,j=7.5hz,2h),7.21–7.15(m,2h),6.91(tt,j=7.3,2.0hz,1h),6.55(t,j=7.0hz,1h),5.32(dq,j=13.6,6.8hz,1h),5.23(dq,j=13.6,6.8hz,1h),3.95(dd,j=11.3,7.1hz,1h),3.60(dd,j=11.3,7.1hz,1h),1.29(dd,j=25.1,6.8hz,12h).13cnmr(126mhz,cdcl3)δ157.9,156.4,155.8,129.8,122.2,117.2,93.5,70.5,44.6,23.3,21.3。
1,3,4-恶二嗪:1hnmr(500mhz,chloroform-d)δ7.36–7.26(m,4h),6.91(tt,j=7.2,2.2hz,1h),6.47(t,j=7.0hz,1h),5.25(h,j=6.8hz,1h),4.34(dd,j=11.5,7.0hz,1h),4.20(dd,j=11.5,6.9hz,1h),4.06(hept,j=6.8hz,1h),1.32(d,j=6.8hz,3h),1.25(dd,j=11.0,6.8hz,6h),1.19(d,j=6.7hz,3h).13cnmr(126mhz,cdcl3)δ156.5,155.6,155.0,129.8,122.2,117.2,90.1,70.5,70.2,63.7,22.0,21.3。
- 还没有人留言评论。精彩留言会获得点赞!