一种黄酮衍生物、其制备方法、用途和包含其的美白产品与流程
本发明属于日化产品技术领域,具体涉及一种黄酮衍生物、其制备方法、用途和包含其的美白产品。
背景技术:
随着社会发展和现代生活节奏加快,由于紫外线、阳光、人体自身等原因使得人体肌肤出现各种问题,较多表现为皮肤细胞损伤,造成皮肤色斑、色素沉积以及干燥等。健康白皙的肌肤是多数人们所追求的,因此具有美白功能的化妆品越来越受到人们的青睐。研究具有美白抗氧化和减少色素沉积等功效的化合物就成了护肤基础研究的重点之一。
目前,美白类化妆品以添加熊果苷、维生素c及其衍生物等具有美白、抗氧化功效的成分为主,但在使用过程中逐渐暴露出吸收差、不稳定、刺激强以及作用效果不明显等问题。所以,开发安全级别高、吸收效果好、稳定性强、性质温和的化妆品原料,成为近年来化妆品研发机构和生产企业关注的热点。
黄酮类化合物是以苯并吡喃为骨架,并且在c2位置有取代基的化合物的总称,包括黄酮、二氢黄酮、黄烷、黄烯等多种结构。基于黄酮类化合物为骨架的天然产物大家庭,具有广泛的生物活性,分布于蔬菜、水果等多种植物中。许多黄酮多酚都具有广泛的的生理功能和活性,如抗氧化、抗肿瘤、抗菌、抗炎、美白等的作用等。儿茶素、柚皮素、橙皮素、青花素、生育酚等天然黄酮多酚化合物一直是护肤品中的重要活性成分。
但是,这些天然黄酮类化合物的美白功效较低,越来越难以满足市场需求,因此具有更强美白功效的产品亟待研发。
技术实现要素:
针对现有技术存在的不足,本发明的目的在于提供一种黄酮衍生物、其制备方法、用途和包含其的美白产品。该黄酮衍生物能够抑制黑色素生成,减少色素沉积,抗氧化,具有美白效果,可用于制备美白产品。
为达此目的,本发明采用以下技术方案:
第一方面,本发明提供一种黄酮衍生物,所述黄酮衍生物具有如下式i或式ii所示结构:
式i、式ii中,r1、r2、r3、r4、r5、r7和r10各自独立地选自氢原子、卤素原子、羟基、氨基、bpin基、取代或未取代的c1-c6的烷基、取代或未取代的c1-c6的烷氧基、取代或未取代的c1-c6的烷氨基、取代或未取代的c2-c6的烯基、取代或未取代的c2-c6的炔基、取代或未取代的c3-c6的环烷基、取代或未取代的c3-c6的环烯基、取代或未取代的c3-c6的环炔基、取代或未取代的c6-c18的芳基、取代或未取代的c5-c18的杂芳基、取代或未取代的c6-c18的芳氧基、取代或未取代的c7-c21的芳烷基中的一种;
r6、r8和r9各自独立地选自氢原子、取代或未取代的c1-c6的烷基、取代或未取代的c2-c6的烯基、取代或未取代的c2-c6的炔基、取代或未取代的c3-c6的环烷基、取代或未取代的c3-c6的环烯基、取代或未取代的c3-c6的环炔基、取代或未取代的c6-c18的芳基、取代或未取代的c5-c18的杂芳基、取代或未取代的c7-c21的芳烷基或
x选自氧原子、硫原子、
如上所述基团中含有取代基时,所述取代基为卤素原子、羟基、羧基、氨基、巯基或硅烷基;
n为0-3(例如0、1、2或3)的整数。
发明人通过研究发现,采用苯乙烯基对黄酮类化合物进行修饰,得到的具有上述结构黄酮衍生物能够显著抑制黑色素生成,减少色素沉积以及抗氧化,具有美白效果,且效果优于未修饰的黄酮类化合物。
本发明中,所述c1-c6的烷基可以是c1、c2、c3、c4、c5或c6的烷基;
所述c1-c6的烷氧基可以是c1、c2、c3、c4、c5或c6的烷氧基;
所述c1-c6的烷氨基可以是c1、c2、c3、c4、c5或c6的烷氨基;
所述c2-c6的烯基可以是c2、c3、c4、c5或c6的烯基;
所述c2-c6的炔基可以是c2、c3、c4、c5或c6的炔基;
所述c3-c6的环烷基可以是c3、c4、c5或c6的环烷基;
所述c3-c6的环烯基可以是c3、c4、c5或c6的环烯基;
所述c3-c6的环炔基可以是c3、c4、c5或c6的环炔基;
所述c6-c18的芳基可以是c6、c7、c10、c12、c14或c18等的芳基;
所述c5-c18的杂芳基可以是c5、c6、c9、c11、c13或c18等的杂芳基;
所述c6-c18的芳氧基可以是c6、c7、c10、c12、c14或c18等的芳氧基;
所述c7-c21的芳烷基可以是c7、c8、c10、c12、c14、c15、c18、c20或c21等的芳烷基。
所述硅烷基优选为四甲基硅基。
需要说明的是,本发明中,具有同一种式i或式ii所示结构的黄酮衍生物还具有不同的构型,包括立体异构体、互变异构体、光学异构体以及对映异构体,不同的异构体之间的性能有所差异。
作为本发明的优选技术方案,所述r1、r2、r3、r4和r5各自独立地选自氢原子、卤素原子、羟基、c1-c6的烷氧基或c6-c18的芳氧基中的一种,且不全为氢原子。
优选地,所述r1、r2、r3、r4和r5各自独立地为氢原子、卤素原子、羟基、甲氧基或乙氧基,且不全为氢原子。
作为本发明的优选技术方案,所述r7为氢原子。
作为本发明的优选技术方案,所述r10选自卤素原子、羟基、氨基、c1-c6的烷基、c1-c6的烷氧基、c1-c6的烷氨基、c2-c6的烯基、c3-c6的环烷基、c2-c6的炔基或bpin基(
优选地,所述r10为卤素原子、羟基、氨基、甲基、甲氧基、乙氨基、乙烯基、环己基、乙炔基或bpin基,n为0-3的整数。
作为本发明的优选技术方案,所述r6、r8和r9各自独立地选自氢原子、c1-c6的烷基、c6-c18的芳基、c7-c21的芳烷基或
优选地,所述r6、r8和r9各自独立地为氢原子、甲基、乙基、苯基、苯甲基或
作为本发明的优选技术方案,所述x为氧原子、硫原子或
优选地,所述r18为氢原子、甲基、乙基或苯甲基。
作为本发明的优选技术方案,所述黄酮衍生物具有如下式iii、式iv、式v、式vi、式vii或式viii所示结构:
式iii-式viii中,r1、r2、r3、r4和r5各自独立地为氢原子、卤素原子、羟基、甲氧基或乙氧基,且不全为氢原子;
r6、r8和r9各自独立地为氢原子、甲基、乙基、苯基、苯甲基或
r10为卤素原子、羟基、氨基、甲基、甲氧基、乙氨基、乙烯基、环己基、乙炔基或bpin基,n为0-3的整数;
x为氧原子、硫原子或
作为本发明的优选技术方案,所述黄酮衍生物选自如下化合物c1-c21中的任意一种或化合物d1-d21中的任意一种:
需要说明的是,上述化合物中的bn为苯甲基(苄基)。
第二方面,本发明提供一种上述黄酮衍生物的制备方法,所述制备方法为:
以黄酮类化合物
其中,r1-r10、x和n具有与本发明第一方面提供的黄酮衍生物的结构式中的r1-r10、x和n相同的含义,在此不再赘述。
作为本发明的优选技术方案,所述黄酮类化合物与苯乙醛类化合物的摩尔比为1:(1.1-5);例如可以是1:1.1、1:1.2、1:1.3、1:1.4、1:1.5、1:1.6、1:1.8、1:2、1:2.2、1:2.5、1:2.8、1:3、1:3.2、1:3.5、1:3.8、1:4、1:4.2、1:4.5、1:4.8或1:5等。
优选地,所述酸性环境中的酸为盐酸、硫酸、磷酸、醋酸或硝酸,更优选为盐酸。
优选地,所述有机溶剂为二乙二醇。
优选地,所述反应的温度为80-140℃,例如可以是80℃、85℃、90℃、95℃、100℃、105℃、110℃、115℃、120℃、125℃、130℃、135℃或140℃等;时间为0.5-12h,例如可以是0.5h、1h、1.5h、2h、2.5h、3h、4h、5h、6h、7h、8h、9h、10h、11h或12h等。
优选地,所述制备方法还包括在反应结束后对反应产物进行提纯,所述提纯的方法为:去除所述有机溶剂,加入乙酸乙酯溶解,水洗,分离有机相,将所述有机相真空浓缩至干,采用柱层析、薄层层析或重结晶的方法分离得到所述黄酮衍生物。
优选地,所述柱层析使用的洗脱液为二氯甲烷和甲醇的混合液。
优选的,二氯甲烷和甲醇的体积比为5:1~80:1,例如可以是5:1、8:1、10:1、12:1、15:1、18:1、20:1、25:1、30:1、35:1、40:1、45:1、50:1、55:1、60:1、65:1、70:1、75:1或80:1等。
第三方面,本发明提供一种第一方面所述的黄酮衍生物的用途,所述黄酮衍生物用于抑制色素生成或抗氧化。
第四方面,本发明提供一种美白产品,所述美白产品含有至少一种本发明第一方面提供的黄酮衍生物。
作为本发明的优选技术方案,所述美白产品为具有抗氧化、淡斑、抗皮肤衰老或抗紫外线功效的护肤品、化妆品、药品或食品。
与现有技术相比,本发明具有以下有益效果:
本发明采用苯乙烯基修饰黄酮类化合物,得到的黄酮衍生物能显著抑制黑色素的生成和抗氧化,其效果优于未修饰的黄酮类化合物及市场常用的α-熊果苷;且本发明提供的黄酮衍生物由于采用纯天然分子进行结构修饰,其安全性好,减低了伤害使用者皮肤的风险,可以用于制备美白用的护肤品、化妆品、药品、保健品或食品等产品。
附图说明
图1为不同化合物在不同浓度下对ros生成的抑制率的柱状图。
具体实施方式
下面结合附图并通过具体实施方式来进一步说明本发明的技术方案。本领域技术人员应该明了,所述具体实施方式仅仅是帮助理解本发明,不应视为对本发明的具体限制。
实施例1
化合物c1(5-苯乙烯基儿茶素)和化合物d1(7-苯乙烯基儿茶素)的制备,具体合成步骤如下:
在100ml圆底烧瓶中,将原料儿茶素(580mg,2mmol,1eq)或表儿茶素(580mg,2mmol,1eq),和苯乙醛(600μl,2.5eq)加入到二乙二醇(40ml)中,滴加5.3%的盐酸(14ml),在120℃下加热反应2小时,反应完毕后,真空除去溶剂,加入乙酸乙酯(50ml)溶解,水洗(20ml×3),分出有机相,有机相真空浓缩至干,使用柱层析分离(选用200-300目的硅胶,洗脱剂为二氯甲烷和甲醇的混合液,体积比20:1),即可得到无色油状目标产物,根据柱层析结果,首先得到c1(5-苯乙烯基儿茶素),收率75%,随后分离得到d1(7-苯乙烯基儿茶素),收率21%。
结构表征:
1hnmr(400mhz,cdcl3),δ=10.27(s,1h),9.48(s,2h),7.72(d,2h),7.44(m,2h),7.30-7.22(m,2h),6.78-6.64(m,4h),5.98(s,1h),5.84(s,1h),5.02(s,1h),4.88-4.86(m,2h),2.81-2.56(m,2h)。
13cnmr(100mhz,cdcl3),δ=166.8,165.7,165.4,156.3,152.1,150.2,136.0,133.1,129.8,129.8,129.8,126.7,125.2,121.1,119.3,66.7,44.5,28.5。
hrms(esi-iontrap)m/z:[m+h]+计算值c23h20o6,329.1360,测定值329.1362。
实施例2
化合物c2的制备,具体合成步骤与实施例1的区别在于,将儿茶素替换为3',4'-二甲氧基儿茶素,收率52%。
结构表征:
1hnmr(400mhz,cdcl3),δ=7.72(m,2h),7.44(m,2h),7.30-7.21(m,2h),6.97-6.96(m,2h),6.87-6.77(m,2h),5.98(s,1h),6.52(d,1h),5.82(s,1h),5.02(s,1h),4.89-4.88(m,2h),3.83(s,3h),3.75(s,3h),2.86-2.81(m,2h)。
hrms(esi-iontrap)m/z:[m+h]+计算值c25h24o6420.1573,测定值420.1572。
实施例3
化合物c3的制备,具体合成步骤如下:
(1)在100ml圆底烧瓶中,将原料阿夫儿茶素(540mg,2mmol,1eq)和氯化亚砜(600μl,2.5eq)加入到1,4-二氧六环(20ml)中,滴加1ml二甲基甲酰胺,室温下反应12小时,反应完毕后,真空除去溶剂,得到4'-氯代阿夫儿茶素;
(2)在100ml圆底烧瓶中,将原料4’-氯代阿夫儿茶素(600mg,1eq)和苯乙醛(600μl,2.5eq)加入到二乙二醇(40ml)中,滴加5.3%的盐酸(14ml),在140℃下加热反应2小时,反应完毕后,真空除去溶剂,加入乙酸乙酯(50ml)溶解,水洗(20ml×3),分出有机相,有机相真空浓缩至干,使用柱层析分离(选用200-300目的硅胶,洗脱剂为二氯甲烷和甲醇的混合液,体积比10:1),即可得到无色油状目标产物,收率78%。
结构表征:
1hnmr(400mhz,cdcl3),δ=9.27(s,1h),7.68(m,2h),7.44-7.40(m,2h),7.38-7.14(m,2h),6.79-6.62(m,4h),6.42(dd,1h),5.99(s,1h),5.87(s,1h),5.04(s,1h),4.96-4.88(m,2h),2.96-2.83(m,2h)。
hrms(esi-iontrap)m/z:[m+h]+计算值c23h17clo6,394.0972,测定值394.0975。
实施例4
化合物c4的制备,具体合成步骤如下:
(1)在100ml圆底烧瓶中,将原料阿夫儿茶素(540mg,2mmol,1eq)加入到草酸(20ml)中,滴加5ml甲醇,100℃反应12小时,反应完毕后,真空除去溶剂,得到2,4,6-三甲氧基阿夫儿茶素;
(2)在100ml圆底烧瓶中,将原料2,4,6-三甲氧基阿夫儿茶素(600mg,1eq)和苯乙醛(600μl,2.5eq)加入到二乙二醇(40ml)中,滴加5.3%的盐酸(14ml),在140℃下加热反应2小时,反应完毕后,真空除去溶剂,加入乙酸乙酯(50ml)溶解,水洗(20ml×3),分出有机相,有机相真空浓缩至干,使用柱层析分离(选用200-300目的硅胶,洗脱剂为二氯甲烷和甲醇的混合液,体积比10:1),即可得到无色油状目标产物,收率82%。
结构表征:
1hnmr(400mhz,cdcl3),δ=9.06(s,1h),7.82-7.81(d,2h),7.65-7.42(m,3h),7.30-7.20(m,2h),6.98-6.54(m,4h),6.20(s,1h),5.08(d,1h),4.56(m,1h),3.78(s,3h),3.72(s,3h),3.41(s,3h),2.84-2.79(m,2h)。
hrms(esi-iontrap)m/z:[m+h]+计算值c26h26o5,418.1780,测定值418.1778。
实施例5
化合物c5的制备,具体合成步骤与实施例1的区别在于,将儿茶素替换为3'-甲氧基-2,4,6三苄氧基儿茶素,收率72%。
结构表征:
1hnmr(400mhz,cdcl3),δ=9.96(s,1h),7.92-7.88(m,4h),7.46-7.39(m,5h),7.35-7.24(m,6h),7.15-7.06(m,6h),6.98-6.84(m,4h),6.78-6.64(m,2h),5.76-5.45(m,4h),5.36-5.11(m,3h),3.37(s,3h),2.79-2.54(m,2h)。
hrms(esi-iontrap)m/z:[m+h]+计算值c45h40o6,676.2825,测定值676.2822。
实施例6
化合物c6的制备,具体合成步骤与实施例1的区别在于,将苯乙醛替换为3,5-二羟基苯乙醛,收率61%。
结构表征:
1hnmr(400mhz,cdcl3),δ=9.65-9.48(m,3h),7.65-7.42(m,3h),7.30-7.20(m,2h),6.98-6.54(m,4h),4.88-4.78(m,2h),2.86-2.61(m,2h)。
hrms(esi-iontrap)m/z:[m+h]+计算值c23h20o8,424.1158,测定值424.1159。
实施例7
化合物c7的制备,具体合成步骤与实施例1的区别在于,将苯乙醛替换为对氨基苯乙醛,收率68%。
结构表征:
1hnmr(400mhz,cdcl3),δ=10.27(s,1h),9.41(s,1h),7.92-7.77(d,2h),7.55-7.41(m,2h),7.30-7.20(m,3h),6.68-6.39(m,3h),4.76-4.66(m,2h),2.97-2.56(m,2h)。
hrms(esi-iontrap)m/z:[m+h]+计算值c23h21no6,407.1369,测定值407.1365。
实施例8
化合物c8的制备,具体合成步骤与实施例1的区别在于,将苯乙醛替换为对乙胺基苯乙醛,收率74%。
结构表征:
1hnmr(400mhz,cdcl3),δ=7.92-7.71(d,2h),7.65-7.52(m,3h),7.38-7.21(m,2h),6.98-6.56(m,3h),4.88-4.85(m,2h),3.45(t,2h),2.81-2.56(m,2h),1.28(t,3h)。
hrms(esi-iontrap)m/z:[m+h]+计算值c25h25no6,435.1682,测定值435.1688。
实施例9
化合物c9的制备,具体合成步骤与实施例1的区别在于,将苯乙醛替换为对溴苯乙醛,收率78%。
结构表征:
1hnmr(400mhz,cdcl3),δ=9.98(s,1h),7.95-7.83(d,2h),7.75-7.52(m,2h),7.33-7.22(m,2h),6.98-6.54(m,4h),4.48-4.20(m,2h),2.81-2.56(m,2h).
hrms(esi-iontrap)m/z:[m+h]+计算值c23h19bro6,470.0365,测定值470.0362。
实施例10
化合物c10的制备,具体合成步骤与实施例1的区别在于,将苯乙醛替换为对甲基苯乙醛,收率69%。
结构表征:
1hnmr(400mhz,cdcl3),δ=9.48(s,1h),7.98-7.88(d,2h),7.77-7.32(m,2h),7.28-7.21(m,3h),6.98-6.64(m,3h),4.90-4.86(m,2h),2.81-2.56(m,2h),2.41(s,3h)。
hrms(esi-iontrap)m/z:[m+h]+计算值c24h22o6,406.1416,测定值406.1418。
实施例11
化合物c11的制备,具体合成步骤与实施例1的区别在于,将苯乙醛替换为对甲氧基苯乙醛,收率55%。
结构表征:
1hnmr(400mhz,cdcl3),δ=10.28(s,1h),7.91-7.83(d,2h),7.77-7.52(m,2h),7.33-7.25(m,2h),6.68-6.54(m,4h),4.90-4.86(m,2h),3.81(s,3h),2.81-2.56(m,2h)。
hrms(esi-iontrap)m/z:[m+h]+计算值c24h22o7,422.1366,测定值422.1371。
实施例12
化合物c12的制备,具体合成步骤与实施例1的区别在于,将苯乙醛替换为对乙炔基苯乙醛,收率63%。
结构表征:
1hnmr(400mhz,cdcl3),δ=10.15(s,1h),9.77(s,1h),7.75-7.73(d,2h),7.65-7.42(m,3h),7.38-7.22(m,2h),6.98-6.54(m,3h),4.88-4.66(m,2h),3.04(s,1h),2.81-2.46(m,2h)。
hrms(esi-iontrap)m/z:[m+h]+计算值c25h20o6,416.1260,测定值416.1266。
实施例13
化合物c13的制备,具体合成步骤与实施例1的区别在于,将苯乙醛替换为对乙烯基苯乙醛,收率75%。
结构表征:
1hnmr(400mhz,cdcl3),δ=9.56(s,1h),7.95-7.83(d,2h),7.79-7.50(m,2h),7.37-7.25(m,3h),6.98-6.54(m,4h),5.76-5.52(m,2h),4.90-4.88(m,2h),2.81-2.56(m,2h)。
hrms(esi-iontrap)m/z:[m+h]+计算值c23h22o6,418.1416,测定值418.1414。
实施例14
化合物c14的制备,具体合成步骤与实施例1的区别在于,将苯乙醛替换为对环己基苯乙醛,收率52%。
结构表征:
1hnmr(400mhz,cdcl3),δ=7.99-7.79(m,2h),7.71-7.52(m,3h),7.34-7.22(m,2h),6.78-6.54(m,2h),5.84(s,1h),4.66-4.62(m,2h),2.80-2.66(m,2h),2.72-2.43(m,3h),2.36-2.31(m,2h),2.11-2.01(m,3h),1.95-1.79(m,3h)。
hrms(esi-iontrap)m/z:[m+h]+计算值c29h30o6,474.2042,测定值474.2043。
实施例15
化合物c15的制备,具体合成步骤如下:
在100ml圆底烧瓶中,将原料egcg(920mg,2mmol,1eq)和苯乙醛(600μl,2.5eq)加入到二乙二醇(40ml)中,滴加5.3%的盐酸(14ml),在140℃下加热反应2小时,反应完毕后,真空除去溶剂,加入乙酸乙酯(50ml)溶解,水洗(20ml×3),分出有机相,有机相真空浓缩至干,使用柱层析分离(选用200-300目的硅胶,洗脱剂为二氯甲烷和甲醇的混合液,体积比10:1),即可得到无色油状目标产物,收率70%。
结构表征:
1hnmr(400mhz,cdcl3),δ=10.42(s,1h),9.31(s,2h),8.73(s,1h),7.78(d,2h),7.45(m,2h),7.20-7.30(m,2h),6.62-6.80(m,4h),5.84-5.98(m,3h),5.02(s,1h),4.86-4.88(m,2h),2.56-2.81(m,2h)。
13cnmr(100mhz,cdcl3),δ=176.8,168.7,165.6,164.2,156.3,152.8,150.2,136.2,133.2,129.6,129.5,129.6,128.7,123.1,119.3,63.7,41.5,29.5。
hrms(esi-iontrap)m/z:[m+h]+计算值c30h24o11,560.1319,测定值560.1321。
实施例16
化合物c16的制备,具体合成步骤与实施例1的区别在于,将儿茶素替换为1-(3',4'-二羟基苯酚)-3h-色烯-2,4,6-三醇,收率52%。
结构表征:
1hnmr(400mhz,cdcl3),δ=10.27(s,1h),7.95-7.83(m,2h),7.71-7.62(m,2h),7.33-7.22(m,3h),6.98-6.74(m,2h),6.64-6,62(m,1h),5.88(m,1h),3.22(m,2h)。
hrms(esi-iontrap)m/z:[m+h]+计算值c23h18o6,390.1103,测定值390.1105。
实施例17
化合物c17的制备,具体合成步骤与实施例1的区别在于,将儿茶素替换为1-(3',4'-二羟基苯酚)-1h-色烯-2,4,6-三醇,收率46%。
结构表征:
1hnmr(400mhz,cdcl3),δ=10.77(s,1h),7.75-7.63(m,2h),7.45-7.32(m,2h),7.33-7.22(m,3h),6.98-6.54(m,4h),5.66(s,1h),5.12(s,1h)。
hrms(esi-iontrap)m/z:[m+h]+计算值c23h18o6,390.1103,测定值390.1107。
实施例18
化合物c18的制备,具体合成步骤与实施例1的区别在于,将儿茶素替换为s-儿茶素,收率72%。
结构表征:
1hnmr(400mhz,cdcl3),δ=9.27(s,1h),7.92-7.71(d,2h),7.65-7.42(m,3h),7.28-7.21(m,3h),6.78-6.56(m,3h),4.26-4.07(m,2h),2.81-2.56(m,2h)。
hrms(esi-iontrap)m/z:[m+h]+计算值c23h20o5s,408.1031,测定值408.1034。
实施例19
化合物c19的制备,具体合成步骤与实施例1的区别在于,将儿茶素替换为n-儿茶素,收率48%。
结构表征:
1hnmr(400mhz,cdcl3),δ=9.48(s,2h),8.29(s,1h),7.72-7.42(m,3h),7.28-7.21(m,3h),6.78-6.61(m,5h),4.31-3.81(m,2h),2.81-2.56(m,2h)。
hrms(esi-iontrap)m/z:[m+h]+计算值c23h21no5,391.1420,测定值391.1422。
实施例20
化合物c20的制备,具体合成步骤如下:
(1)在100ml圆底烧瓶中,将原料n-儿茶素(540mg,1eq)加入到二氯甲烷(20ml)中,缓慢加入氢化钠(1g),随后加入乙醇(5ml),室温反应12小时,反应完毕后,真空除去溶剂,得到n-乙基儿茶素;
(2)在100ml圆底烧瓶中,将原料n-乙基儿茶素(600mg,1eq)和苯乙醛(600μl,2.5eq)加入到二乙二醇(40ml)中,滴加5.3%的盐酸(14ml),在140℃下加热反应2小时,反应完毕后,真空除去溶剂,加入乙酸乙酯(50ml)溶解,水洗(20ml×3),分出有机相,有机相真空浓缩至干,使用柱层析分离(选用200-300目的硅胶,洗脱剂为二氯甲烷和甲醇的混合液,体积比10:1),即可得到无色油状目标产物,收率56%。
结构表征:
1hnmr(400mhz,cdcl3),δ=9.27(s,1h),7.64-7.42(m,3h),7.29-7.12(m,3h),6.78-6.64(m,2h),6.35-6.24(m,3h),4.31(m,1h),3.81(m,1h),3.39(t,2h),2.81-2.56(s,2h),1.12(t,3h).
hrms(esi-iontrap)m/z:[m+h]+计算值c25h25no5,329.1360,测定值329.1362。
实施例21
化合物c21的制备,具体合成步骤如下:
(1)在100ml圆底烧瓶中,将原料n-儿茶素(540mg,1eq)加入到二氯甲烷(20ml)中,缓慢加入氢化钠(1g),随后加入溴化苄(5ml),室温反应12小时,反应完毕后,真空除去溶剂,得到n-苄基儿茶素;
(2)在100ml圆底烧瓶中,将原料n-苄基儿茶素(600mg,1eq)和苯乙醛(600μl,2.5eq)加入到二乙二醇(40ml)中,滴加5.3%的盐酸(14ml),在140℃下加热反应2小时,反应完毕后,真空除去溶剂,加入乙酸乙酯(50ml)溶解,水洗(20ml×3),分出有机相,有机相真空浓缩至干,使用柱层析分离(选用200-300目的硅胶,洗脱剂为二氯甲烷和甲醇的混合液,体积比10:1),即可得到无色油状目标产物,收率69%。
结构表征:
1hnmr(400mhz,cdcl3),δ=7.92-7.71(d,3h),7.65-7.52(m,3h),7.44(s,1h),7.38-7.21(m,2h),6.98-6.56(m,3h),6.49-6.41(m,3h),6.22(s,1h),5.37(s,1h),4.61.4-45(m,2h),3.81(s,1h),2.81-2.55(m,2h)。
hrms(esi-iontrap)m/z:[m+h]+计算值c30h27no5,481.1889,测定值481.1888。
实施例22
化合物c1(5-苯乙烯基表儿茶素)的制备,具体合成步骤与实施例1的区别在于,将儿茶素替换为表儿茶素,收率55%。
结构表征:
1hnmr(400mhz,cdcl3),δ=10.27(s,1h),9.48(s,1h),7.85(d,2h),7.54-7.45(m,2h),7.30-7.22(m,2h),6.88-6.74(m,4h),5.86(s,1h),5.57(s,1h),5.02(s,1h),4.88-4.77(m,2h),2.95-2.85(m,2h)。
13cnmr(100mhz,cdcl3),δ=166.8,165.7,165.4,156.3,152.1,150.2,136.0,133.1,129.8,129.8,129.8,126.7,125.2,121.1,119.3,66.7,44.5,28.5。
hrms(esi-iontrap)m/z:[m+h]+计算值c23h20o6,329.1360,测定值329.1362。
需要说明的是,实施例1提供的化合物c1(5-苯乙烯基儿茶素)和实施例22提供的化合物c1(5-苯乙烯基表儿茶素)为化合物c1的两种不同的构型,二者互为异构体,因此加以区分。其他实施例中未对化合物的构型进行说明,其具体的构型本领域技术人员可根据制备其的原料的构型进行判断。
本发明实施例提供的化合物中的活性氢(-oh)在1hnmr测试中可能表征不出,因此部分化合物通过1hnmr积分得到的氢原子数量小于其理论氢原子数量。
本发明实施例中采用的化合物的结构式如下:
对上述实施例提供的化合物进行如下性能测试:
1、细胞毒性实验
将上述实施例制备得到的化合物,以及儿茶素、egcg、α-熊果苷分别用dmso配制成不同浓度的水溶性试剂,备用。
把b16-f10细胞(小鼠黑色素瘤细胞)按每孔20000枚的密度接种在96孔板上,培养基为加入10wt%fbs(血清)和1wt%双抗的1640培养基。在接种b16-f10细胞的96孔板上每孔加入90μl培养基,然后在每孔中分别加入10μl上述不同浓度的水溶性试剂(以dmso作为空白对照,α-熊果苷作为阳性对照),每一种化合物均设置5个浓度梯度,终浓度分别为6.25μm、12.5μm、25μm、50μm、100μm,然后在37℃培养箱中培养2天。
之后向每孔加入10μlcck-8溶液,将96孔板在37℃培养箱中培养4小时。用酶标仪测定在450nm处的吸光度,并计算对b16-f10细胞的抑制率。抑制率=[1-(od加药-od本底)/(od空白-od本底)]×100%,其中od加药为实验组的吸光度,od空白为空白组的吸光度,od本底为本底的吸光度。绘制抑制率-浓度标准曲线,根据曲线公式计算出各化合物的ic50值(抑制率为50%时的药物浓度)。
ic50值如下表1所示:
表1
表1中,“∞”表示该化合物的毒性极低,无法根据前述公式计算出ic50值。
从表1结果可以看出,儿茶素、表儿茶素、egcg以及α-熊果苷对于b16-f10(小鼠黑色素瘤细胞)的毒性都非常小,本发明提供的黄酮类衍生物也具有较低的毒性。
2、黑色素生成抑制实验:
将上述实施例制备得到的化合物,以及儿茶素、egcg、α-熊果苷分别用dmso配制成不同浓度的水溶性试剂,备用。
(1)把b16-f10细胞(小鼠黑色素瘤细胞)按每孔20000枚的密度接种在96孔板上,培养基为加入10wt%fbs(血清)和1wt%双抗的1640培养基。在接种b16-f10细胞的96孔板上每孔加入90μl培养基和10μlα-msh(粗黑色素细胞激素),然后在每孔中分别加入10μl上述不同浓度的水溶性试剂(以dmso作为空白对照,α-熊果苷作为阳性对照),每一种化合物均设置5个浓度梯度,终浓度分别为6.25μm、12.5μm、25μm、50μm、100μm;以不添加α-msh且水溶性试剂为dmso的组作为本底;然后在37℃培养箱中培养2天。
培养结束后,利用酶标仪在405nm的波长下测量培养液的吸光度,并计算对黑色素生成的抑制率。抑制率=[1-(od加药-od本底)/(od空白-od本底)]×100%,其中od加药为实验组的吸光度,od空白为空白组的吸光度,od本底为本底的吸光度。
抑制率数据如下表2所示:
表2
(2)把melan-a细胞(小鼠黑色素细胞)按每孔20000枚的密度接种在96孔板上,培养基为加入10wt%fbs(血清)和1wt%双抗的1640培养基。在接种melan-a细胞的96孔板上每孔加入90μl的培养基和10μlα-msh(粗黑色素细胞激素),在每孔中分别加入10μl上述不同浓度的水溶性试剂(以dmso作为空白对照,α-熊果苷作为阳性对照),每一种化合物均设置5个浓度梯度,终浓度分别为6.25μm、12.5μm、25μm、50μm、100μm;以不添加α-msh且水溶性试剂为dmso的组作为本底;然后在37℃培养箱中培养2天。
培养结束后,利用酶标仪在405nm的波长下测量培养液的吸光度,并计算对黑色素生成的抑制率。抑制率=[1-(od加药-od本底)/(od空白-od本底)]×100%,其中od加药为实验组的吸光度,od空白为空白组的吸光度,od本底为本底的吸光度。
抑制率数据如下表3所示:
表3
由表2和表3的数据可以看出,相较于未经苯乙烯基修饰的黄酮类化合物,本发明提供的黄酮衍生物对黑色素生成的抑制率得到了显著提升。
3、细胞氧化应激能力抑制实验
把人原发性表皮角质细胞按每孔15000枚的密度接种在96孔板上,培养基为加入10wt%fbs和1wt%双抗的1640培养基。在接种melan-a细胞的96孔板上每孔加入90μl的培养基,在37℃培养箱中培养24小时。在每孔分别加入10μl上述不同浓度的水溶性试剂(以dmso作为空白对照,α-熊果苷作为阳性对照),每一种化合物均设置5个浓度梯度,终浓度分别为6.25μm、12.5μm、25μm、50μm、100μm。随后每孔加入20μl,200μm的h2o2溶液(以不添加h2o2溶液的空白对照组作为本底),在37℃培养箱中培养2小时,随后向每孔加入100μl含有25μmdcfa(2,7-二氢二氯荧光黄)的pbs(磷酸盐缓冲液),在37℃培养箱中培养0.5小时。
培养结束后,利用酶标仪在485nm波长和535nm波长下测量培养液的吸光度,并计算对ros(活性氧簇)生成的抑制率。抑制率=[1-(od加药-od本底)/(od空白-od本底)]×100%,其中od加药为实验组的吸光度,od空白为空白组的吸光度,od本底为本底的吸光度。
抑制率数据如下表4所示:
表4
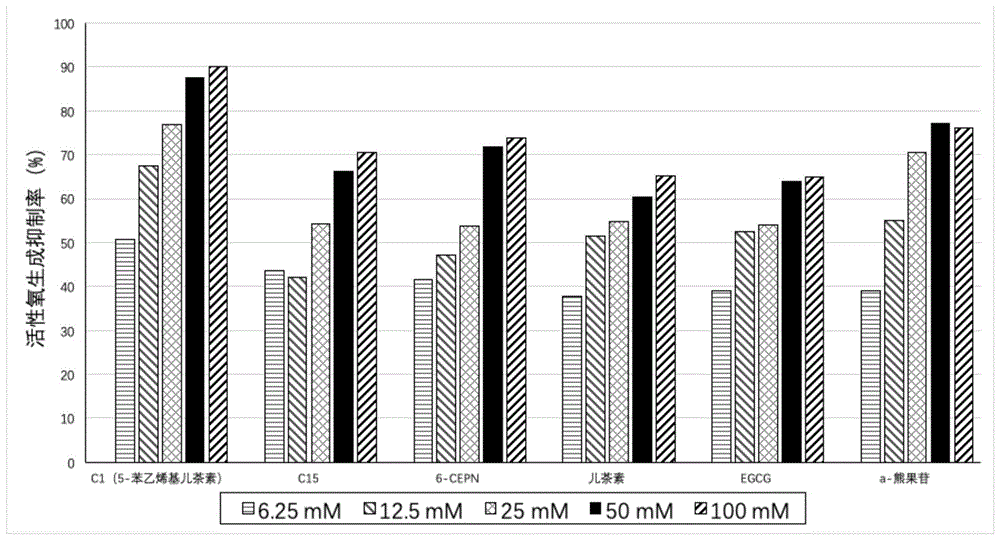
其中,化合物c1(5-苯乙烯基儿茶素)、c15、6-cepn、儿茶素、egcg和α-熊果苷在不同浓度下对ros生成的抑制率的柱状图如图1所示。
由图1和表4的结果可以看出,相较于未经苯乙烯基修饰的黄酮类化合物,本发明提供的黄酮衍生物抗氧化功能得到了显著提升。
申请人声明,以上所述仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,所属技术领域的技术人员应该明了,任何属于本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到的变化或替换,均落在本发明的保护范围和公开范围之内。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种1‑(4‑氯苯基)‑2‑吡啶‑1‑基‑1‑基乙酮溴化物的制备方法与制造工艺
- 一种1‑(4‑溴苯基)‑2‑吡啶‑1‑基‑1‑基乙酮溴化物的制备方法与制造工艺
- 一类噁唑并香豆素衍生物在农药方面的应用
- 一种香豆素衍生物、其制备方法以及由其制得的水凝胶的制作方法
- 3-(2-芳基-1h-吲哚-3-基)-4-羟基香豆素衍生物及其合成方法
- 一种羟甲香豆素的药物组合物及其医药用图
- 含卤素或苯并咪唑类功能基团的香豆素类衍生物及其制备方法和应用
- 一种有机叠氮与香豆素合成3-氨基苯并吡喃-2-酮类衍生物的新方法
- 香豆素-3-羧酸/1-辛烷磺酸/LGdH复合体及其合成方法
- 香豆素-3-羧酸/1-辛烷磺酸/LGdH复合体及其合成方法