一种从黄连中提取吡喃葡萄糖苷衍生物的工艺的制作方法
[0001]
本发明属于化学提纯领域,具体涉及一种从黄连中提取吡喃葡萄糖苷衍生物的工艺。
背景技术:
[0002]
黄连为毛茛科植物黄连(coptis chinensisfranch.)(味连)的干燥根茎。味连,又名川连、鸡爪黄连,主产四川,湖北、湖南、陕西、甘肃等省亦有栽培。味苦、性寒,归心、脾、胃、肝、胆、大肠经。具有清热燥湿,泻火解毒的功效。主要用于湿热痞满、呕吐吞酸、泻痢、黄疸、高热神昏、心火亢盛、心烦不寐、血热、目赤、牙痛、痈肿疔疮,外治湿疹、湿疮、耳道流脓。
[0003]
文献“黄连水提液化学成分的分离与鉴定,李雪改等”公开了一种从黄连水提液中分离化合物的方法,并提取鉴定了22个化合物,其提取分离方法如下:黄连干燥根茎1kg,用水回流提取2次(第一次8倍量,第二次6倍量),每次2h,合并提取液,48℃下减压回收溶剂,浓缩后得到总浸膏(1l0 g)。采用重结晶、反复开放硅胶柱色谱、sephadex lh一20柱色谱、制备薄层色谱以及phplc等手段,对黄连的水提液进行系统分离,共得到22个化合物,并鉴定了它们的结构。得到的22个化合物中,包括一种吡喃葡萄糖苷衍生物,即其化合物15:3-(3’,4’二羟基苯基)-(2r)-乳酸-4
’-
o-β-d-吡喃葡萄糖苷(3-(3’,4'-dihydroxyphe
·
ny1)-(2r)-lactic acid-4
’-
o-β-d-glucopyranoside)。
[0004][0005]
本申请人申请的中国专利201610459981.5中公开了上述化合物15(即其式iv所示化合物)在制备用于预防和/或治疗糖尿病药物中的用途,该化合物的10mg/kg,20mg/kg及40mg/kg三个剂量组对正常小鼠糖耐量试验中给葡萄糖后60min血糖均有显著的降低作用,20mg/kg及40mg/kg还能降低auc并升高血糖抑制率,且未见明显毒副作用,药物安全有效。因此,上述式iv所示化合物在制备用于预防和/或治疗糖尿病药物中具有非常好的应用前景。
[0006]
但是,该化合物的制备方法非常复杂,成本高,收率低,不适合商业化生产。因此,寻找一种简单耗时短、操作简单、收率高的方法来制得上述化合物,具有非常好的应用前景。
技术实现要素:
[0007]
本发明的目的在于提供一种从黄连中提取式iv所示化合物的工艺。
[0008]
本发明提供了一种式iv所示化合物的制备方法,所述方法包括以下步骤:
[0009]
1)将黄连干燥、粉碎,得到药材粉末;
[0010]
2)将步骤1)所得药材粉末用提取溶剂提取,得到提取液;所述提取溶剂选自水,醇类溶剂,或水和醇类溶剂的混合溶液;
[0011]
3)将步骤2)所得提取液浓缩,加入酸,酸化,析出固体,然后过滤,去除固体,保留滤液;
[0012]
4)取步骤3)所得滤液,采用层析柱,以乙醇体积百分数为0-80%的水溶液为洗脱剂进行梯度洗脱,收集乙醇体积百分数为20%-60%时的洗脱液a;
[0013]
5)将步骤4)所得洗脱液a浓缩,得浓缩液,采用层析柱,以乙醇体积百分数为0-80%的水溶液为洗脱剂对浓缩液进行梯度洗脱,收集乙醇体积百分数为30%-40%的洗脱液b,浓缩,干燥,即得;
[0014]
其中,式iv所示化合物的结构为:
[0015]
进一步地,步骤4)中,收集的是乙醇体积百分数为40%的洗脱液a;步骤5)中,收集的是乙醇体积百分数为30%的洗脱液b。
[0016]
进一步地,步骤1)中,所述干燥方式包括阴干、阳干、烘干,干燥后产物中水分重量在14%以下;所述粉碎为粉碎至10-80目;
[0017]
优选地,所述干燥方式为在以下条件下烘干:在40-200℃下干燥0.5-10小时。
[0018]
进一步地,步骤2)中,所述提取方式选自回流提取、超声提取、浸泡提取中的一种或两种以上;其中,所述浸泡提取的温度为60-90℃;
[0019]
和/或,所述药材粉末与提取溶剂的质量比为1:(2-10),优选为1:(6-8);
[0020]
和/或,所述提取次数为2-3次,每次的提取时间为1.5-2h;
[0021]
和/或,所述提取溶剂选自水或体积浓度为30%的乙醇。
[0022]
进一步地,步骤3)中,所述酸选自盐酸、硫酸、醋酸的一种或两种以上,酸化后体系的ph值为1.5-6.5;
[0023]
和/或,浓缩后体系的体积为浓缩前的15%。
[0024]
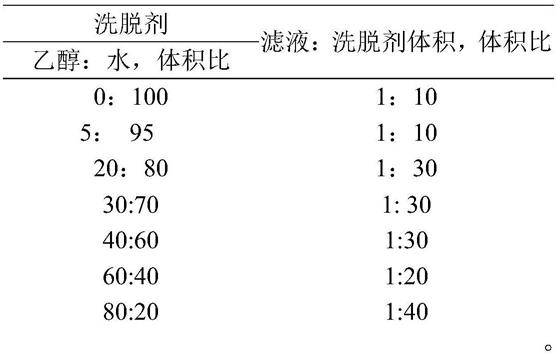
进一步地,步骤4)中,所述梯度洗脱的条件如下:
[0025][0026]
进一步地,步骤5)中,所述梯度洗脱的条件如下:
[0027][0028]
进一步地,步骤4)中,所述层析柱选自hpd-400树脂柱、d-101树脂柱、hpd-100树脂柱、mcl树脂柱、ab-8树脂柱、ods柱;优选为ods柱或mcl树脂柱;
[0029]
步骤5)中,所述层析柱的填料选自mcl树脂柱、ods柱、hpd-300树脂柱、d-301树脂柱、x-5树脂柱;优选为ods柱。
[0030]
进一步地,步骤5)中,所述干燥的方式选自喷雾干燥、真空干燥、冷冻干燥、近红外干燥以及微波干燥中的一种或两种以上,干燥温度为70℃以下;
[0031]
和/或,浓缩液的体积为洗脱液a的5%。
[0032]
进一步地,步骤1)中,所述黄连选自黄连花、黄连叶、黄连根、黄连茎和黄连须中的一种或两种以上;优选为黄连根或黄连叶。
[0033]
ods柱即十八烷基硅烷键合硅胶柱。
[0034]
实验结果表明,本发明提供的方法能够从黄连中提取得到式iv所示化合物,本方法操作简单、工艺流程短耗时少,层析柱填料可重复利用,生产总成本降低,产品收率、纯度高,适合商业化生产。
[0035]
本发明中,阴干指在没有阳光直射、通风性很好的地方自然干燥;阳干指在阳光直射下干燥;烘干指加热干燥。
[0036]
显然,根据本发明的上述内容,按照本领域的普通技术知识和惯用手段,在不脱离
本发明上述基本技术思想前提下,还可以做出其它多种形式的修改、替换或变更。
[0037]
以下通过实施例形式的具体实施方式,对本发明的上述内容再作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术均属于本发明的范围。
具体实施方式
[0038]
本发明所用原料与设备均为已知产品,通过购买市售产品所得。
[0039]
实施例1、本发明的提取工艺
[0040]
1、提取
[0041]
(1)将黄连的提取部位黄连根干燥,在40-200℃,干燥0.5-10小时,至水分14%以下,然后粉碎10-80目,得到药材粉末;
[0042]
(2)将步骤(1)所得药材粉末中加入6倍量水,在60hz下超声提取2次,每次提取1小时,得到提取液;
[0043]
2、提纯
[0044]
(3)将步骤2)所得提取液浓缩,加入酸,酸化,析出固体,然后过滤,去除固体,保留滤液;
[0045]
(4)取步骤3)所得滤液,采用hpd-400树脂柱,以乙醇体积百分数为0-80%的水溶液为洗脱剂进行梯度洗脱,收集乙醇体积百分数为30%时的洗脱液a;所述梯度洗脱的条件如下:
[0046][0047]
(5)将步骤4)所得洗脱液a浓缩,得浓缩液,采用mcl树脂柱,以乙醇体积百分数为0-80%的水溶液为洗脱剂对浓缩液进行梯度洗脱,收集乙醇体积百分数为40%的洗脱液b,浓缩,得浸膏,将浸膏干燥,即得式iv所示化合物:
[0048]
[0049]
所述梯度洗脱的条件如下:
[0050][0051]
3、表征
[0052]
通过核磁检测与对照品核磁图谱比较,确认为iv所示化合物。通过色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以水为流动相b,hplc检测分析纯度为91%;称量干燥后化合物计算得出收率为36%。
[0053]
实施例2、本发明的提取工艺
[0054]
1、提取
[0055]
(1)将黄连的提取部位黄连根干燥,在40-200℃,干燥0.5-10小时,至水分14%以下,然后粉碎10-80目,得到药材粉末;
[0056]
(2)将步骤(1)所得药材粉末中加8倍量30%乙醇水溶液,在80℃下回流提取2次,每次提取1.5小时,得到提取液;
[0057]
2、提纯
[0058]
(3)将步骤2)所得提取液浓缩,加入酸,酸化,析出固体,然后过滤,去除固体,保留滤液;
[0059]
(4)取步骤3)所得滤液,采用d-101树脂柱,以乙醇体积百分数为0-80%的水溶液为洗脱剂进行梯度洗脱,收集乙醇体积百分数为20%的洗脱液a,所述梯度洗脱的条件与实施例1的步骤(4)相同;
[0060]
(5)将步骤4)所得洗脱液a浓缩,得浓缩液,采用ods柱,以乙醇体积百分数为0-80%的水溶液为洗脱剂对浓缩液进行梯度洗脱,收集乙醇体积百分数为30%的洗脱液b(所述梯度洗脱的条件与实施例1的步骤(5)相同),浓缩,得浸膏,将浸膏干燥,即得式iv所示化合物。
[0061]
3、表征
[0062]
通过核磁检测与对照品核磁图谱比较,确认为iv所示化合物。通过色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以水为流动相b,hplc检测分析纯度为92%;称量干燥后化合物重量计算得出收率为35%。
[0063]
实施例3、本发明的提取工艺
[0064]
1、提取
[0065]
(1)将黄连的提取部位黄连根干燥,在40-200℃,干燥0.5-10小时,至水分14%以下,然后粉碎10-80目,得到药材粉末;
[0066]
(2)将步骤(1)所得药材粉末中加入6倍量水,在90℃下浸泡提取3次,每次提取1小时,得到提取液;
[0067]
2、提纯
[0068]
(3)将步骤2)所得提取液浓缩,加入酸,酸化,析出固体,然后过滤,去除固体,保留滤液;
[0069]
(4)取步骤3)所得滤液,采用hpd-100树脂柱,以乙醇体积百分数为0-80%的水溶液为洗脱剂进行梯度洗脱,收集乙醇体积百分数为20%的洗脱液a,所述梯度洗脱的条件与实施例1的步骤(4)相同;
[0070]
(5)将步骤4)所得洗脱液a浓缩,得浓缩液,采用hpd-300树脂柱,以乙醇体积百分数为0-80%的水溶液为洗脱剂对浓缩液进行梯度洗脱,收集乙醇体积百分数为30%的洗脱液b(所述梯度洗脱的条件与实施例1的步骤(5)相同),浓缩,得浸膏,将浸膏干燥,即得式iv所示化合物。
[0071]
3、表征
[0072]
通过核磁检测与对照品核磁图谱比较,确认为iv所示化合物。通过色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以水为流动相b,hplc检测分析纯度为88%;称量干燥后化合物计算得出收率为34%。
[0073]
实施例4、本发明的提取工艺
[0074]
1、提取
[0075]
(1)将黄连的提取部位黄连根干燥,在40-200℃,干燥0.5-10小时,至水分14%以下,然后粉碎10-80目,得到药材粉末;
[0076]
(2)将步骤(1)所得药材粉末中加入8倍量水,在70℃下浸泡提取2次,每次提取2小时,得到提取液;
[0077]
2、提纯
[0078]
(3)将步骤2)所得提取液浓缩,加入酸,酸化,析出固体,然后过滤,去除固体,保留滤液;
[0079]
(4)取步骤3)所得滤液,采用hpd-400树脂柱,以乙醇体积百分数为0-80%的水溶液为洗脱剂进行梯度洗脱,收集乙醇体积百分数为20%的洗脱液a,所述梯度洗脱的条件与实施例1的步骤(4)相同;
[0080]
(5)将步骤4)所得洗脱液a浓缩,得浓缩液,采用mcl树脂柱,以乙醇体积百分数为0-80%的水溶液为洗脱剂对浓缩液进行梯度洗脱,收集乙醇体积百分数为40%的洗脱液b(所述梯度洗脱的条件与实施例1的步骤(5)相同),浓缩,得浸膏,将浸膏干燥,即得式iv所示化合物。
[0081]
3、表征
[0082]
通过核磁检测与对照品核磁图谱比较,确认为iv所示化合物。通过色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以水为流动相b,hplc检测分析纯度为90%;称量干燥后化合物计算得出收率为32%。
[0083]
实施例5、本发明的提取工艺
[0084]
1、提取
[0085]
(1)将黄连的提取部位黄连叶干燥,在40-200℃,干燥0.5-10小时,至水分14%以下,然后粉碎10-80目,得到药材粉末;
[0086]
(2)将步骤(1)所得药材粉末中加入6倍量水,在80℃下浸泡提取2次,每次提取2小时,得到提取液;
[0087]
2、提纯
[0088]
(3)将步骤2)所得提取液浓缩,加入酸,酸化,析出固体,然后过滤,去除固体,保留滤液;
[0089]
(4)取步骤3)所得滤液,采用mcl树脂柱,以乙醇体积百分数为0-80%的水溶液为洗脱剂进行梯度洗脱,收集乙醇体积百分数为60%的洗脱液a,所述梯度洗脱的条件与实施例1的步骤(4)相同;
[0090]
(5)将步骤4)所得洗脱液a浓缩,得浓缩液,采用d-301树脂柱,以乙醇体积百分数为0-80%的水溶液为洗脱剂对浓缩液进行梯度洗脱,收集乙醇体积百分数为30%的洗脱液b(所述梯度洗脱的条件与实施例1的步骤(5)相同),浓缩,得浸膏,将浸膏干燥,即得式iv所示化合物。
[0091]
3、表征
[0092]
通过核磁检测与对照品核磁图谱比较,确认为iv所示化合物。通过色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以水为流动相b,hplc检测分析纯度为80%;称量干燥后化合物计算得出收率为15%。
[0093]
实施例6、本发明的提取工艺
[0094]
1、提取
[0095]
(1)将黄连的提取部位黄连根干燥,在40-200℃,干燥0.5-10小时,至水分14%以下,然后粉碎10-80目,得到药材粉末;
[0096]
(2)将步骤(1)所得药材粉末中加入6倍量水,在70℃下浸泡提取2次,每次提取2小时,得到提取液;
[0097]
2、提纯
[0098]
(3)将步骤2)所得提取液浓缩,加入酸,酸化,析出固体,然后过滤,去除固体,保留滤液;
[0099]
(4)取步骤3)所得滤液,采用ab-8树脂柱,以乙醇体积百分数为0-80%的水溶液为洗脱剂进行梯度洗脱,收集乙醇体积百分数为30%的洗脱液a,所述梯度洗脱的条件与实施例1的步骤(4)相同;
[0100]
(5)将步骤4)所得洗脱液a浓缩,得浓缩液,采用x-5树脂柱,以乙醇体积百分数为0-80%的水溶液为洗脱剂对浓缩液进行梯度洗脱,收集乙醇体积百分数为30%的洗脱液b(所述梯度洗脱的条件与实施例1的步骤(5)相同),浓缩,得浸膏,将浸膏干燥,即得式iv所示化合物。
[0101]
3、表征
[0102]
通过核磁检测与对照品核磁图谱比较,确认为iv所示化合物。通过色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以水为流动相b,hplc检测分析纯度为85%;称量干燥后化合物计算得出收率为28%。
[0103]
实施例7、本发明的提取工艺
[0104]
1、提取
[0105]
(1)将黄连的提取部位黄连根干燥,在40-200℃,干燥0.5-10小时,至水分14%以下,然后粉碎10-80目,得到药材粉末;
[0106]
(2)将步骤(1)所得药材粉末中加入6倍量30%乙醇水溶液,在70℃下回流提取2次,每次提取1.5小时,得到提取液;
[0107]
2、提纯
[0108]
(3)将步骤2)所得提取液浓缩,加入酸,酸化,析出固体,然后过滤,去除固体,保留滤液;
[0109]
(4)取步骤3)所得滤液,采用ods柱,以乙醇体积百分数为0-80%的水溶液为洗脱剂进行梯度洗脱,收集乙醇体积百分数为40%的洗脱液a,所述梯度洗脱的条件与实施例1的步骤(4)相同;(5)将步骤4)所得洗脱液a浓缩,得浓缩液,采用ods柱,以乙醇体积百分数为0-80%的水溶液为洗脱剂对浓缩液进行梯度洗脱,收集乙醇体积百分数为30%的洗脱液b(所述梯度洗脱的条件与实施例1的步骤(5)相同),浓缩,得浸膏,将浸膏干燥,即得式iv所示化合物。
[0110]
3、表征
[0111]
通过核磁检测与对照品核磁图谱比较,确认为iv所示化合物。通过色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以水为流动相b,hplc检测分析纯度为94%;称量干燥后化合物计算得出收率为38%。
[0112]
实施例8、本发明的提取工艺
[0113]
1、提取
[0114]
(1)将黄连的提取部位黄连根干燥,在40-200℃,干燥0.5-10小时,至水分14%以下,然后粉碎10-80目,得到药材粉末;
[0115]
(2)将步骤(1)所得药材粉末中加入8倍量30%乙醇水溶液,在80℃下回流提取2次,每次提取2小时,得到提取液;
[0116]
2、提纯
[0117]
(3)将步骤2)所得提取液浓缩,加入酸,酸化,析出固体,然后过滤,去除固体,保留滤液;
[0118]
(4)取步骤3)所得滤液,采用mcl柱,以乙醇体积百分数为0-80%的水溶液为洗脱剂进行梯度洗脱,收集乙醇体积百分数为40%的洗脱液a,所述梯度洗脱的条件与实施例1的步骤(4)相同;
[0119]
(5)将步骤4)所得洗脱液a浓缩,得浓缩液,采用ods柱,以乙醇体积百分数为0-80%的水溶液为洗脱剂对浓缩液进行梯度洗脱,收集乙醇体积百分数为30%的洗脱液b(所述梯度洗脱的条件与实施例1的步骤(5)相同),浓缩,得浸膏,将浸膏干燥,即得式iv所示化合物。
[0120]
3、表征
[0121]
通过核磁检测与对照品核磁图谱比较,确认为iv所示化合物。通过色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以水为流动相b,hplc检测分析纯度为92%;称量干燥后化合物计算得出收率为40%。
[0122]
实施例9、本发明的提取工艺
[0123]
1、提取
[0124]
(1)将黄连的提取部位黄连根干燥,在40-200℃,干燥0.5-10小时,至水分14%以下,然后粉碎10-80目,得到药材粉末;
[0125]
(2)将步骤(1)所得药材粉末中加入8倍量,在60hz下回流提取3次,每次提取1小时,得到提取液;
[0126]
2、提纯
[0127]
(3)将步骤2)所得提取液浓缩,加入酸,酸化,析出固体,然后过滤,去除固体,保留滤液;
[0128]
(4)取步骤3)所得滤液,采用ods柱,以乙醇体积百分数为0-80%的水溶液为洗脱剂进行梯度洗脱,收集乙醇体积百分数为30%的洗脱液a,所述梯度洗脱的条件与实施例1的步骤(4)相同;
[0129]
(5)将步骤4)所得洗脱液a浓缩,得浓缩液,mcl树脂柱,以乙醇体积百分数为0-80%的水溶液为洗脱剂对浓缩液进行梯度洗脱,收集乙醇体积百分数为40%的洗脱液b(所述梯度洗脱的条件与实施例1的步骤(5)相同),浓缩,得浸膏,将浸膏干燥,即得式iv所示化合物。
[0130]
3、表征
[0131]
通过核磁检测与对照品核磁图谱比较,确认为iv所示化合物。通过色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以水为流动相b,hplc检测分析纯度为90%;计算原药材中iv所示化合物含量,称量干燥后化合物计算得出收率为38%。
[0132]
实施例10、本发明的提取工艺
[0133]
1、提取
[0134]
(1)将黄连的提取部位黄连根干燥,在40-200℃,干燥0.5-10小时,至水分14%以下,然后粉碎10-80目,得到药材粉末;
[0135]
(2)将步骤(1)所得药材粉末中加入6倍量水,在90℃下浸泡提取2次,每次提取2小时,得到提取液;
[0136]
2、提纯
[0137]
(3)将步骤2)所得提取液浓缩,加入酸,酸化,析出固体,然后过滤,去除固体,保留滤液;
[0138]
(4)取步骤3)所得滤液,采用mcl树脂柱,以乙醇体积百分数为0-80%的水溶液为洗脱剂进行梯度洗脱,收集乙醇体积百分数为40%的洗脱液a,所述梯度洗脱的条件与实施例1的步骤(4)相同;
[0139]
(5)将步骤4)所得洗脱液a浓缩,得浓缩液,采用mcl树脂柱,以乙醇体积百分数为0-80%的水溶液为洗脱剂对浓缩液进行梯度洗脱,收集乙醇体积百分数为40%的洗脱液b(所述梯度洗脱的条件与实施例1的步骤(5)相同),浓缩,得浸膏,将浸膏干燥,即得式iv所示化合物。
[0140]
3、表征
[0141]
通过核磁检测与对照品核磁图谱比较,确认为iv所示化合物。通过色谱条件与系
统适用性试验以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以水为流动相b,hplc检测分析纯度为90%;称量干燥后化合物计算得出收率为33%。
[0142]
表1各样品的提取工艺及收率、纯度对比
[0143][0144][0145]
从表1可以看出,采用本发明实施例1-10的方法均能够从黄连中提取得到式iv所示化合物。特别是采用实施例7或8的方法,能够得到高收率、高纯度的目标产物,即采用回流提取的方式、提取两次后,再以乙醇体积百分数为0-80%的水溶液为洗脱剂,依次通过2次ods柱进行梯度洗脱,或者依次通过mcl树脂柱和ods柱进行梯度洗脱,分别收集乙醇体积百分数为40%和30%的洗脱液,得到的目标产物的收率高达38%-40%,纯度高达92%-94%。
[0146]
综上,实验结果表明,本发明提供的方法能够从黄连中提取得到式iv所示化合物,本方法操作简单、层析柱填料可重复利用,使总体成本降低,产品收率高、纯度高,适合商业化生产。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1