一种罗库溴铵中间体及罗库溴铵的制备方法与流程
本发明涉及制药领域,具体涉及一种罗库溴铵中间体及罗库溴铵的制备方法。
背景技术:
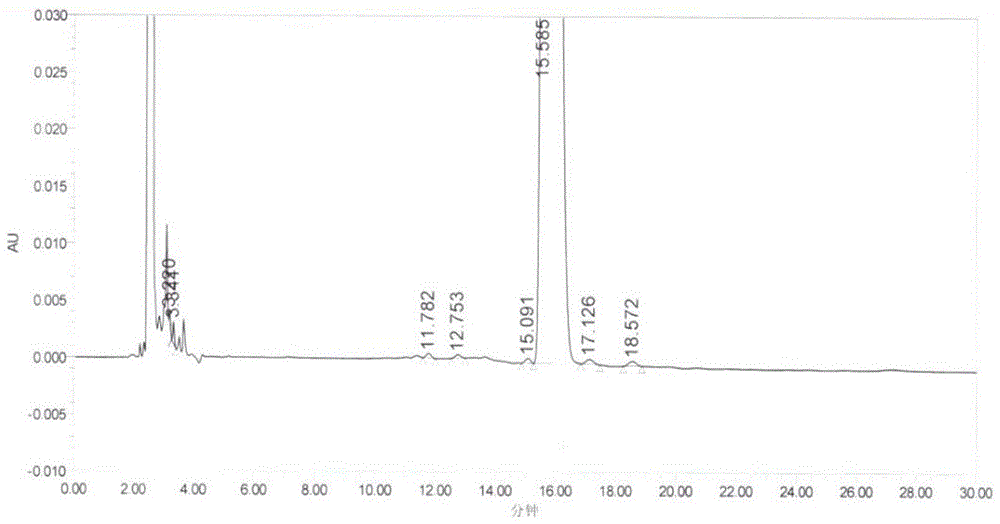
:罗库溴铵(rocuroniumbromide)是一种新型的非去极化甾醇类肌松药,由荷兰欧加农(organon)公司研究开发,于1994年6月在美国首次上市。本品作为麻醉辅助用药,用于麻醉时的气管插管和手术时的肌肉松弛,具有起效快,持续时间短,恢复迅速等特点。该药是目前国际上应用最广泛的肌松药。目前罗库溴铵常用的合成方法是以市售式ⅱ化合物为起始原料,经采用常规的酯化方式得到式ⅲ化合物,式ⅲ化合物再与3-溴丙烯季铵化反应得到。专利us4894369中,描述了以式ⅱ化合物为起始原料,以强腐蚀性试剂乙酰氯为酯化试剂,得到式ⅲ化合物粗品,经柱层析得到化合物3精品,收率47.8%,然后再与3-溴丙烯成盐得到罗库溴铵。该方法后处理繁琐,收率低,且使用了腐蚀性较大的乙酰氯,对环境不友好。专利wo2009016648中提到以乙酰咪唑为酯化试剂,25℃反应48小时,然后将反应液用水洗涤除去杂质,然后干燥、浓缩有机相,得到式ⅲ化合物粗品,经两次重结晶得到高纯度式ⅲ化合物(纯度>99.9%),再与3-溴丙烯25℃反应24小时,得到成品罗库溴铵。该方法反应时间长,后处理反复用水洗涤,增加了式ⅲ化合物降解的风险。专利us7569687用乙酰氯或乙酸酐在室温反应22小时,反应完毕后用碱中和,有机层用水洗,再用硫酸钠干燥,滤液浓缩加乙腈精制,再用卤代烃/乙腈混合溶剂精制得到式ⅲ化合物,然后再与3-溴丙烯,在碳酸钠催化下制备得到罗库溴铵。该方法式ⅲ化合物制备后处理涉及酸碱中和,水洗,硫酸钠干燥等,工业废水量大,操作复杂。专利cn101125878中以式ⅵ化合物为原料,在酸性条件下经选择性降解3位乙酰氧得到式ⅲ化合物,然后再与3-溴丙烯成盐制备得到罗库溴铵。该方法后处理操作麻烦,涉及大量酸性废水,有较大的环境污染风险。因此,选用一种后处理简单的新工艺制备罗库溴铵中间体及罗库溴铵是有必要的。技术实现要素:本发明的目的:针对现有技术的不足,提供一种罗库溴铵中间体及罗库溴铵制备方法:这种制备方法后处理简单,适用于大规模工业化应用。本发明采用以下技术方案:一种罗库溴铵中间体及罗库溴铵的制备方法,其特征在于,包括以下步骤:s1:以(2β,3α,5α,16β,17β)-2-(4-吗啉)-16-(1-吡咯)-雄甾-3,17-二醇(式ⅱ)为原料,在缩合剂催化下与冰醋酸经过乙酰化得化合物(2β,3α,5α,16β,17β)-2-(4-吗啉)-16-(1-吡咯)-雄甾-3,17-二醇-17-醋酸酯(式ⅲ);s2:将s1所得化合物(2β,3α,5α,16β,17β)-2-(4-吗啉)-16-(1-吡咯)-雄甾-3,17-二醇-17-醋酸酯(式ⅲ)在碱性催化剂作用下与3-溴丙烯反应即得罗库溴铵(式ⅰ);反应式如下:进一步的,一种罗库溴铵中间体及罗库溴铵的制备方法,其特征在于,包括以下步骤:s1:以(2β,3α,5α,16β,17β)-2-(4-吗啉)-16-(1-吡咯)-雄甾-3,17-二醇(式ⅱ)为原料,在缩合剂催化下与冰醋酸经过乙酰化反应,然后冷却,析晶得化合物(2β,3α,5α,16β,17β)-2-(4-吗啉)-16-(1-吡咯)-雄甾-3,17-二醇-17-醋酸酯(式ⅲ);s2:将s1所得化合物(2β,3α,5α,16β,17β)-2-(4-吗啉)-16-(1-吡咯)-雄甾-3,17-二醇-17-醋酸酯(式ⅲ)在碱性催化剂作用下与3-溴丙烯反应,析晶,然后在氮气保护下过滤后干燥,即得罗库溴铵(式ⅰ);反应式如下:进一步的,所述步骤s1中使用的溶剂为二氯甲烷、丙酮、乙酸乙酯、甲苯、乙腈、n,n-二甲基甲酰胺、二甲亚砜中的一种或几种;优选的,所述溶剂为乙腈;进一步的,所述步骤s1中使用的缩合剂为喹啉类型缩合剂2-乙氧基-1-乙氧碳酰基-1,2-二氢喹啉(式ⅳ化合物,缩写为eedq),结构如下所示:或1-异丁氧基甲酰基-2-异丁氧基-1,2-二氢喹啉(式ⅴ化合物,缩写为iidq),结构如下:优选的,所述缩合剂为2-乙氧基-1-乙氧碳酰基-1,2-二氢喹啉;进一步的,所述步骤s1中反应温度为0℃~100℃;优选的,所述反应温度为80~90℃;进一步优选的,所述反应温度为82℃;进一步的,所述步骤s1中,式ⅱ化合物:缩合剂:冰醋酸三者物质的量之比为1:1~10.0:1~10.0;优选的,式ⅱ化合物:缩合剂:冰醋酸三者物质的量之比为1:4.5:4.0;进一步的,所述步骤s1的反应时间为2~24h;优选的,所述步骤s1的反应时间为4~5h;进一步的,所述步骤s1的冷却温度为-10~30℃;优选的,所述冷却温度为-5~5℃;进一步的,所述步骤s2的溶剂为质子性溶剂;进一步优选的,所述溶剂为甲醇、乙醇、异丙醇,正丁醇、叔丁醇中的一种或几种;优选的,溶剂为异丙醇;进一步的,所述步骤s2中的碱性催化剂为碳酸氢钠、碳酸钠、碳酸钾、氧化铝中的一种或几种;优选的,碱性催化剂为氧化铝;进一步的,所述步骤s2中的3-溴丙烯与(2β,3α,5α,16β,17β)-2-(4-吗啉)-16-(1-吡咯)-雄甾-3,17-二醇-17-醋酸酯(式ⅲ)的物质的量的之比为4~10:1;优选的,所述3-溴丙烯与(2β,3α,5α,16β,17β)-2-(4-吗啉)-16-(1-吡咯)-雄甾-3,17-二醇-17-醋酸酯(式ⅲ)的物质的量的之比为5:1;进一步的,所述步骤s2的反应时间为4~7h;优选的,所述步骤s2的反应时间为6h;进一步的,所述步骤s2的反应温度为25~40℃;优选的,所述反应温度为25~30℃;进一步的,所述步骤s2的干燥方式为减压干燥;优选的,所述干燥温度为30~50℃;进一步优选40℃。附图说明图1为实施例4制备得到式ⅰ化合物的hplc图谱图2为实施例4制备得到式ⅲ化合物的hplc图谱有益效果1.本发明采用新型喹啉类型缩合剂催化乙酰化反应,高选择性与17位羟基反应,避免副反应发生,减少3位酰化杂质生成,所得的产物纯度较高。2.本发明反应后处理简单,不使用酸性水、碱性水对产物洗涤,不会产生大量酸性或碱性废水,降低环境污染风险。具体实施方式实施例1:向干燥、洁净的250ml三口瓶中,加入10g式ⅱ化合物,加入150ml甲苯,30.6iidq,5.4g冰醋酸,升温至回流反应5小时。降温至10~20℃,析出晶体,过滤、干燥得化合物(式ⅲ)7.1g,摩尔收率64.9%,纯度>99.9%。向干燥、洁净的100ml三口瓶中,加入7g式ⅲ化合物,50ml无水乙醇,0.5g碳酸钠,7g3-溴丙烯,35℃反应4小时,减压过滤除去碳酸钠,滤液缓慢滴入至700ml甲基叔丁醚中,搅拌析晶,氮气保护下过滤,40℃减压干燥过夜,得罗库溴铵(式ⅰ)7g,摩尔收率80.1%,纯度>99.0%。实施例2向干燥、洁净的250ml三口瓶中,加入10g式ⅱ化合物(2β,3α,5α,16β,17β)-2-(4-吗啉)-16-(1-吡咯)-雄甾-3,17-二醇,加入150ml甲苯,30.6giidq,5.4g冰醋酸,升温至回流反应5小时。降温至10~20℃,析出晶体,过滤、干燥得化合物(式ⅲ)6.8g,摩尔收率62.2%,纯度>99.9%;向干燥、洁净的100ml三口瓶中,加入6g式ⅲ化合物(2β,3α,5α,16β,17β)-2-(4-吗啉)-16-(1-吡咯)-雄甾-3,17-二醇-17-醋酸酯,50ml无水乙醇,0.5g碳酸钠,6g3-溴丙烯,35℃反应4小时,减压过滤除去碳酸钠,滤液缓慢滴入至700ml甲基叔丁醚中,搅拌析晶,氮气保护下过滤,40℃减压干燥过夜,得罗库溴铵(式ⅰ)5.9g,摩尔收率78.8%,纯度>99.0%。实施例3:向干燥、洁净的2000ml三口瓶中,加入50g式ⅱ化合物,加入0.5l丙酮,124.6geedq,26.9g冰醋酸,升温至回流反应4小时,加入0.75l乙腈。然后缓慢降温至室温,保温析晶2小时,过滤,得白色结晶粉末(式ⅲ)48g,摩尔收率87.7%,纯度>99.9%;向干燥、洁净的1000ml三口瓶中,加入45g式ⅲ化合物,900ml叔丁醇,10g氧化铝,50g3-溴丙烯,45℃反应6小时,减压过滤除去氧化铝,滤液缓慢滴入至5l无水乙醚中,搅拌析晶,氮气保护下过滤,40℃减压干燥过夜,得罗库溴铵(式ⅰ)50g,摩尔收率89.3%,纯度>99.0%。实施例4:向干燥、洁净的1000ml三口瓶中,加入50g式ⅱ化合物,加入800ml乙腈,124.6geedq,26.9g冰醋酸,升温至回流反应4.5小时。反应完毕降温至-5℃保温搅拌3小时,过滤,得到白色结晶固体(式ⅲ)50g,摩尔收率91.4%,纯度>99.9%。向干燥、洁净的500ml三口瓶中,加入40g式ⅲ化合物,200ml异丙醇,5g氧化铝,60g3-溴丙烯,25~30℃反应6小时,减压过滤除去氧化铝,滤液缓慢滴入至2000ml乙酸乙酯中,搅拌析晶,氮气保护下过滤,减压干燥得罗库溴铵48(式ⅰ)g,收率96.1%,纯度>99.0%。实验例5对实施例4获得的式ⅲ化合物以及式ⅰ化合物罗库溴铵进行hplc色谱分析,测得式ⅲ化合物tr=3.310min,其纯度>99.9%(如图2所示),式ⅰ化合物罗库溴铵tr=15.585min,其纯度>99.6%(如图1所示),由此可见,本发明制备出的产物纯度极高。其中表示化合物纯度的色谱峰面积积分结果分别如下:(1)式ⅲ化合物色谱峰结果(2)式ⅰ化合物色谱峰结果编号保留时间tr面积峰高面积%115.5851163208849465099.67当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1