新型反应性海藻酸衍生物的制作方法
本发明涉及海藻酸衍生物。
背景技术:
海藻酸是从巨藻(レッソニア)、巨囊藻(マクロシスティス)、海带(ラミナリア)、泡叶藻(アスコフィラム)、丛梗藻(ダービリア)、褐藻苷苔(eckloniacava)、黑海带(アラメ)、昆布等褐藻类中提取的生物体内吸收性的多糖类,是d-甘露糖醛酸(m)和l-葡糖醛酸(g)这2种糖醛酸聚合为直链状的聚合物。更具体而言,是由d-甘露糖醛酸的均聚物组分(mm组分)、l-葡糖醛酸的均聚物组分(gg组分)、和d-甘露糖醛酸和l-葡糖醛酸随机配列而得的组分(m/g组分)任意键合而得的嵌段共聚物。
这样的海藻酸在食品、医疗、化妆品、纤维、造纸等广泛的领域中应用。
在此,进行了将海藻酸改变为适应各用途的物质尝试(专利文献1~3)。此外,已知记载了马来酰亚胺和/或硫醇作为交联基团的多糖类衍生物(专利文献4~7)。
现有技术文献
专利文献
专利文献1:日本特开2010-209130号公报
专利文献2:日本特开2007-99902号公报
专利文献3:国际公开第2004/099259号
专利文献4:日本特表2003-516519号公报
专利文献5:日本特表2015-502957号公报
专利文献6:fr2967678号
专利文献7:国际公开第2014/058359号。
技术实现要素:
发明要解决的问题
上述情况下,需求新的海藻酸衍生物。
用于解决问题的手段
本发明人等为了解决上述课题进行了深入研究,结果发现,导入了规定的交联基团的规定的海藻酸衍生物的交联后的稳定性得到改善等,从而完成本发明。
即,本发明如以下方式[1-1]~[22b]所述。
[1-1]海藻酸衍生物,其中,在选自海藻酸、它的酯和它的盐中至少1种的羧基的一部分上具有下述式(i)所示的基团(式中,虚线右侧除外):
[化1]
(式中,
-a1-为选自下述式中的连接基(式中,两虚线外侧除外):
[化2]
r1各自独立地为选自氢原子、甲基、异丙基、异丁基、仲丁基、羟基甲基、2-羟基乙基、硫醇甲基、甲基硫基乙基、羧基甲基、羧基乙基、氨基羰基甲基、氨基羰基乙基、氨基丁基、胍基丙基、苯甲基、4-羟基苯甲基、3-吲哚基甲基、4-咪唑基甲基、和与r1所键合的碳原子和该碳原子所键合的氮原子一起形成环的丙烷-1,3-二基中的基团;r2各自独立地为选自氢原子、甲基、异丙基、异丁基、仲丁基、羟基甲基、2-羟基乙基、硫醇甲基、甲基硫基乙基、羧基甲基、羧基乙基、氨基羰基甲基、氨基羰基乙基、氨基丁基、胍基丙基、苯甲基、4-羟基苯甲基、3-吲哚基甲基、4-咪唑基甲基、和与r2所键合的碳原子和该碳原子所键合的氮原子一起形成环的丙烷-1,3-二基中的基团;
n为1~18的整数;
m为1~9的整数;
j为0~9的整数)。
[1-2]根据前述[1-1]所述的海藻酸衍生物,其中,式(i)中,-a1-为选自下述式中的连接基(式中,两虚线外侧除外):
[化3]
n为1~18的整数;
m为1~9的整数。
[2]根据前述[1-1]或[1-2]所述的海藻酸衍生物,其中,式(i)中,-a1-为选自下述式中的连接基(式中,两虚线外侧除外):
[化4]
[3]根据前述[1-1]~[2]中任一项所述的海藻酸衍生物,其中,式(i)所示的基团选自下述式(式中,虚线右侧除外):
[化5]
[4]根据前述[1-1]~[3]中任一项所述的海藻酸衍生物,其中,交联基团的导入率为1%~30%。
[5]根据前述[1-1]~[4]中任一项所述的海藻酸衍生物,其中,海藻酸衍生物的利用凝胶过滤色谱法测定得到的重均分子量为10万da~300万da。
[1a-1]海藻酸衍生物,其中,在选自海藻酸、它的酯和它的盐中至少1种的羧基的一部分上具有下述式(i)所示的基团(式中,虚线右侧除外):
[化6]
(式中,-a1-为选自下述式中的连接基(式中,两虚线外侧除外):
[化7]
r1各自独立地为选自氢原子、甲基、异丙基、异丁基、仲丁基、羟基甲基、2-羟基乙基、硫醇甲基、甲基硫基乙基、羧基甲基、羧基乙基、氨基羰基甲基、氨基羰基乙基、氨基丁基、胍基丙基、苯甲基、4-羟基苯甲基、3-吲哚基甲基、4-咪唑基甲基和与r1所键合的碳原子和该碳原子所键合的氮原子一起形成环的丙烷-1,3-二基中的基团;
r2各自独立地为选自氢原子、甲基、异丙基、异丁基、仲丁基、羟基甲基、2-羟基乙基、硫醇甲基、甲基硫基乙基、羧基甲基、羧基乙基、氨基羰基甲基、氨基羰基乙基、氨基丁基、胍基丙基、苯甲基、4-羟基苯甲基、3-吲哚基甲基、4-咪唑基甲基和与r2所键合的碳原子和该碳原子所键合的氮原子一起形成环的丙烷-1,3-二基中的基团;
n为1~18的整数;
m为1~9的整数;
j为0~9的整数)。
[1a-2]根据前述[1a-1]所述的海藻酸衍生物,其中,式(i)中,-a1-为选自下述式中的连接基(式中,两虚线外侧除外):
[化8]
n为1~18的整数;
m为1~9的整数;
j为0~9的整数。
[2a]根据前述[1a-1]或[1a-2]所述的海藻酸衍生物,其中,式(i)中,-a1-为选自下述式中的连接基(式中,两虚线外侧除外):
[化9]
[2a-1]前述方式[2a]中,优选为-a1-为选自下述式中的连接基(式中,两虚线外侧除外):
[化10]
[3a]根据上述[1a-1]~[2a]中任一项所述的海藻酸衍生物,其中,式(i)所示的基团选自下述式(式中,虚线右侧除外):
[化11]
[3a-1]前述方式[3a]中,优选为式(i)所示的基团为选自下述式中的基团(式中,虚线右侧除外):
[化12]
[4a]根据前述[1a-1]~[3a-1]中任一项所述的海藻酸衍生物,其中,式(i)所示的基团的导入率为1%~30%。
[5a]根据前述[1a-1]~[4a]中任一项所述的海藻酸衍生物,其中,海藻酸衍生物的利用凝胶过滤色谱法测定得到的重均分子量为10万da~300万da。
[1b-1]海藻酸衍生物,其中,在选自海藻酸、它的酯和它的盐中至少1种的羧基的一部分上具有下述式(i)所示的基团(式中,虚线右侧除外):
[化13]
(式中,-a1-为选自下述式中的连接基(式中,两虚线外侧除外):
[化14]
r1各自独立地为选自氢原子、甲基、异丙基、异丁基、仲丁基、羟基甲基、2-羟基乙基、硫醇甲基、甲基硫基乙基、羧基甲基、羧基乙基、氨基羰基甲基、氨基羰基乙基、氨基丁基、胍基丙基、苯甲基、4-羟基苯甲基、3-吲哚基甲基、4-咪唑基甲基和与r1所键合的碳原子和该碳原子所键合的氮原子一起形成环的丙烷-1,3-二基中的基团;
r2各自独立地为选自氢原子、甲基、异丙基、异丁基、仲丁基、羟基甲基、2-羟基乙基、硫醇甲基、甲基硫基乙基、羧基甲基、羧基乙基、氨基羰基甲基、氨基羰基乙基、氨基丁基、胍基丙基、苯甲基、4-羟基苯甲基、3-吲哚基甲基、4-咪唑基甲基和与r2所键合的碳原子和该碳原子所键合的氮原子一起形成环的丙烷-1,3-二基中的基团;
n为1~18的整数;
m为1~9的整数;
j为0~9的整数)
(其中,-a1-=-ch2ch2-除外)。
[1b-2]根据前述[1b-1]所述的海藻酸衍生物,其中,式(i)中,-a1-为选自下述式中的连接基(式中,两虚线外侧除外):
[化15]
n为1~18的整数;
m为1~9的整数;
j为0~9的整数)
(其中,-a1-=-ch2ch2-除外)。
[2b]根据前述[1b-1]或[1b-2]所述的海藻酸衍生物,其中,式(i)中,-a1-为选自下述式中的连接基(式中,两虚线外侧除外):
[化16]
[2b-1]前述方式[2b]中,优选为-a1-为选自下述式中的连接基(式中,两虚线外侧除外):
[化17]
[3b]根据上述[1b-1]~[2b]中任一项所述的海藻酸衍生物,其中,式(i)所示的基团选自下述式(式中,虚线右侧除外):
[化18]
[3b-1]前述方式[3b]中,优选为式(i)所示的基团为选自下述式中的基团(式中,虚线右侧除外):
[化19]
[4b]根据前述[1b-1]~[3b-1]中任一项所述的海藻酸衍生物,其中,式(i)所示的基的导入率为1%~30%。
[5b]根据前述[1b-1]~[4b]中任一项所述的海藻酸衍生物,其中,海藻酸衍生物的利用凝胶过滤色谱法测定得到的重均分子量为10万da~300万da。
[6]海藻酸衍生物,其中,在选自海藻酸、它的酯和它的盐中至少1种的羧基的一部分上具有下述式(ii)所示的交联基团(式中,虚线右侧除外):
[化20]
(式中,
p1为氢原子或硫醇基(-sh基)的保护基,
-a2-为下述式所示的连接基(式中,两波浪线外侧除外):
[化21]
前述-a2-中,ar为可被水溶性取代基(例如,1~2个)取代的亚苯基;
n4为0~10的整数;
m4为0~10的整数;
p为0~10的整数)。
[7]根据前述[6]所述的海藻酸衍生物,其中,式(ii)中,p1为氢原子、乙酰基或苯甲酰基。
[7-1]根据前述[6]所述的海藻酸衍生物,其中,式(ii)中,p1为氢原子、或乙酰基。
[8]根据前述[6]~[7-1]中任一项所述的海藻酸衍生物,其中,式(ii)的-a2-中,ar为对亚苯基。
[8-1]根据前述[6]~[7-1]中任一项所述的海藻酸衍生物,其中,-a2-为选自下述式中的连接基(各式中,两虚线外侧除外):
[化22]
ar为对亚苯基。
[9]根据前述[6]~[8-1]中任一项所述的海藻酸衍生物,其中,式(ii)所示的基团选自下述式(式中,虚线右侧除外):
[化23]
[9-1]根据前述[6]~[8-1]中任一项所述的海藻酸衍生物,其中,式(ii)所示的基团选自下述式(各式中,虚线右侧除外):
[化24]
[10]根据前述[6]~[9-1]中任一项所述的海藻酸衍生物,其中,式(ii)所示的基团的导入率(有时称为“交联基团的导入率”)为1%~30%。
[11]根据上述[6]~[10]中任一项所述的海藻酸衍生物,其中,海藻酸衍生物的利用凝胶过滤色谱法测定得到的重均分子量为10万da~300万da。
[12]组合物,其含有前述[1-1]~[5]中任一项所述的海藻酸衍生物和前述[6]~[11]中任一项所述的海藻酸衍生物。
[12a]组合物,其含有前述[1a-1]~[5a]中任一项所述的海藻酸衍生物和前述[6]~[11]中任一项所述的海藻酸衍生物。
[12b]组合物,其含有前述[1b-1]~[5b]中任一项所述的海藻酸衍生物和前述[6]~[11]中任一项所述的海藻酸衍生物。
[13-1]交联海藻酸结构体,其通过对前述[1-1]~[5]中任一项所述的海藻酸衍生物和前述[6]~[11]中任一项所述的海藻酸衍生物实施交联反应而得到。
[13a-1]交联海藻酸结构体,其通过对前述[1a-1]~[5a]中任一项所述的海藻酸衍生物和前述[6]~[11]中任一项所述的海藻酸衍生物实施交联反应而得到。
[13b-1]交联海藻酸结构体,其通过对前述[1b-1]~[5b]中任一项所述的海藻酸衍生物和前述[6]~[11]中任一项所述的海藻酸衍生物实施交联反应而得到。
[13-2]根据前述[13-1]所述的交联海藻酸结构体,其中,交联海藻酸结构体通过将前述[1-1]~[5]中任一项所述的海藻酸衍生物的溶液滴加在包含钙离子的溶液中,对所得凝胶在前述[6]~[11]中任一项所述的海藻酸衍生物的溶液中实施交联反应而得到。
[13a-2]根据前述[13a-1]所述的交联海藻酸结构体,其中,交联海藻酸结构体通过将前述[1a-1]~[5a]中任一项所述的海藻酸衍生物的溶液滴加在包含钙离子的溶液中,对所得凝胶在前述[6]~[11]中任一项所述的海藻酸衍生物的溶液中实施交联反应而得到。
[13b-2]根据前述[13b-1]所述的交联海藻酸结构体,其中,交联海藻酸结构体通过将前述[1b-1]~[5b]中任一项所述的海藻酸衍生物的溶液滴加在包含钙离子的溶液中,对所得凝胶在前述[6]~[11]中任一项所述的海藻酸衍生物的溶液中实施交联反应而得到。
[13-3]根据前述[13-1]所述的交联海藻酸结构体,其中,交联海藻酸结构体通过将前述[6]~[11]中任一项所述的海藻酸衍生物的溶液滴加在包含钙离子的溶液中,对所得凝胶在前述[1-1]~[5]中任一项所述的海藻酸衍生物的溶液中实施交联反应而得到。
[13a-3]根据前述[13a-1]所述的交联海藻酸结构体,其中,交联海藻酸结构体通过将前述[6]~[11]中任一项所述的海藻酸衍生物的溶液滴加在包含钙离子的溶液中,对所得凝胶在前述[1a-1]~[5a]中任一项所述的海藻酸衍生物的溶液中实施交联反应而得到。
[13b-3]根据前述[13b-1]所述的交联海藻酸结构体,其中,交联海藻酸结构体通过将前述[6]~[11]中任一项所述的海藻酸衍生物的溶液滴加在包含钙离子的溶液中,对所得凝胶在前述[1b-1]~[5b]中任一项所述的海藻酸衍生物的溶液中实施交联反应而得到。
[13-4]根据前述[13-1]所述的交联海藻酸结构体,其中,交联海藻酸结构体通过将前述[12]所述的组合物的溶液滴加在包含钙离子的溶液中而得到。
[13a-4]根据前述[13a-1]所述的交联海藻酸结构体,其中,交联海藻酸结构体通过将前述[12a]所述的组合物的溶液滴加在包含钙离子的溶液中而得到。
[13b-4]根据前述[13b-1]所述的交联海藻酸结构体,其中,交联海藻酸结构体通过将前述[12b]所述的组合物的溶液滴加在包含钙离子的溶液中而得到。
[13-5]根据前述[13-1]~[13-4]中任一项所述的交联海藻酸结构体,其中,交联海藻酸结构体为纤维、珠、近球形的凝胶或微囊。
[13a-5]根据前述[13a-1]~[13a-4]中任一项所述的交联海藻酸结构体,其中,交联海藻酸结构体为纤维、珠、近球形的凝胶或微囊。
[13b-5]根据前述[13b-1]~[13b-4]中任一项所述的交联海藻酸结构体,其中,交联海藻酸结构体为纤维、珠、近球形的凝胶或微囊。
[14]医疗用材料,其包含前述[13-1]~[13-5]中任一项所述的交联海藻酸结构体。
[14a]医疗用材料,其包含前述[13a-1]~[13a-5]中任一项所述的交联海藻酸结构体。
[14b]医疗用材料,其包含前述[13b-1]~[13b-5]中任一项所述的交联海藻酸结构体。
[15]根据前述[14]所述的医疗用材料,其中,交联海藻酸结构体为珠或近球形的凝胶。
[15a]根据前述[14a]所述的医疗用材料,其中,交联海藻酸结构体为珠或近球形的凝胶。
[15b]根据前述[14b]所述的医疗用材料,其中,交联海藻酸结构体为珠或近球形的凝胶。
[16]交联海藻酸结构体的制造方法,其包括:将前述[1-1]~[5]中任一项所述的海藻酸衍生物的溶液滴加在包含钙离子的溶液中,对所得凝胶在前述[6]~[11]中任一项所述的海藻酸衍生物的溶液中实施交联反应。
[16a]交联海藻酸结构体的制造方法,其包括:将前述[1a-1]~[5a]中任一项所述的海藻酸衍生物的溶液滴加在包含钙离子的溶液中,对所得凝胶在前述[6]~[11]中任一项所述的海藻酸衍生物的溶液中实施交联反应。
[16b]交联海藻酸结构体的制造方法,其包括:将前述[1b-1]~[5b]中任一项所述的海藻酸衍生物的溶液滴加在包含钙离子的溶液中,对所得凝胶在前述[6]~[11]中任一项所述的海藻酸衍生物的溶液中实施交联反应。
[17]交联海藻酸结构体的制造方法,其包括:将前述[6]~[11]中任一项所述的海藻酸衍生物的溶液滴加在包含钙离子的溶液中,对所得凝胶在前述[1-1]~[5]中任一项所述的海藻酸衍生物的溶液中实施交联反应。
[17a]交联海藻酸结构体的制造方法,其包括:将前述[6]~[11]中任一项所述的海藻酸衍生物的溶液滴加在包含钙离子的溶液中,对所得凝胶在前述[1a-1]~[5a]中任一项所述的海藻酸衍生物的溶液中实施交联反应。
[17b]交联海藻酸结构体的制造方法,其包括:将前述[6]~[11]中任一项所述的海藻酸衍生物的溶液滴加在包含钙离子的溶液中,对所得凝胶在前述[1b-1]~[5b]中任一项所述的海藻酸衍生物的溶液中实施交联反应。
[18]交联海藻酸结构体的制造方法,其包括:将前述[12]所述的组合物的溶液滴加在包含钙离子的溶液中。
[18a]交联海藻酸结构体的制造方法,其包括:将前述[12a]所述的组合物的溶液滴加在包含钙离子的溶液中。
[18b]交联海藻酸结构体的制造方法,其包括:将前述[12b]所述的组合物的溶液滴加在包含钙离子的溶液中。
[19]交联海藻酸结构体的制造方法,其包括:将前述[1-1]~[5]中任一项所述的海藻酸衍生物利用2价金属离子部分地进行交联,得到特定的结构体后,利用前述[6]~[11]中任一项所述的海藻酸衍生物实施交联反应。
[19a]交联海藻酸结构体的制造方法,其包括:将前述[1a-1]~[5a]中任一项所述的海藻酸衍生物利用2价金属离子部分地进行交联,得到特定的结构体后,利用前述[6]~[11]中任一项所述的海藻酸衍生物实施交联反应。
[19b]交联海藻酸结构体的制造方法,其包括:将前述[1b-1]~[5b]中任一项所述的海藻酸衍生物利用2价金属离子部分地进行交联,得到特定的结构体后,利用前述[6]~[11]中任一项所述的海藻酸衍生物实施交联反应。
[20]交联海藻酸结构体的制造方法,其包括:将前述[6]~[11]中任一项所述的海藻酸衍生物利用2价金属离子部分地进行交联,得到特定的结构体后,利用前述[1-1]~[5]中任一项所述的海藻酸衍生物实施交联反应。
[20a]交联海藻酸结构体的制造方法,其包括:将前述[6]~[11]中任一项所述的海藻酸衍生物利用2价金属离子部分地进行交联,得到特定的结构体后,利用前述[1a-1]~[5a]中任一项所述的海藻酸衍生物实施交联反应。
[20b]交联海藻酸结构体的制造方法,其包括:将前述[6]~[11]中任一项所述的海藻酸衍生物利用2价金属离子部分地进行交联,得到特定的结构体后,利用前述[1b-1]~[5b]中任一项所述的海藻酸衍生物实施交联反应。
[21]交联海藻酸结构体的制造方法,其包括:将前述[12]所述的组合物利用2价金属离子部分地进行交联。
[21a]交联海藻酸结构体的制造方法,其包括:将前述[12a]所述的组合物利用2价金属离子部分地进行交联。
[21b]交联海藻酸结构体的制造方法,其包括:将前述[12b]所述的组合物利用2价金属离子部分地进行交联。
[22]交联海藻酸结构体,其通过实施包括使用前述[1-1]~[5]中任一项所述的海藻酸衍生物、前述[6]~[11]中任一项所述的海藻酸衍生物、和2价金属离子的交联反应而得到,具有内容物的保持性。
[22a]交联海藻酸结构体,其通过实施包括使用前述[1a-1]~[5a]中任一项所述的海藻酸衍生物、前述[6]~[11]中任一项所述的海藻酸衍生物、和2价金属离子的交联反应而得到,具有内容物的保持性。
[22b]交联海藻酸结构体,其通过实施包括使用前述[1b-1]~[5b]中任一项所述的海藻酸衍生物、前述[6]~[11]中任一项所述的海藻酸衍生物、和2价金属离子的交联反应而得到,具有内容物的保持性。
发明效果
本发明提供新的海藻酸衍生物。优选海藻酸衍生物的交联后的稳定性得到改善。
附图说明
[图1]显示交联海藻酸结构体(alg-2、al-ex-2/al-ex-7-1、或al-ex-3/al-ex-7-1)的凝胶稳定性的评价的图。
[图2]显示交联海藻酸结构体(al-2、al-ex-2/al-ex-7-1、或al-ex-3/al-ex-7-1)的凝胶泄漏率的评价的图。
[图3]显示交联海藻酸结构体(al-ex-8/al-ex-7-1-2、al-ex-9/al-ex-7-1-2、或al-ex-10/al-ex-7-1-2)的凝胶稳定性的评价的图。
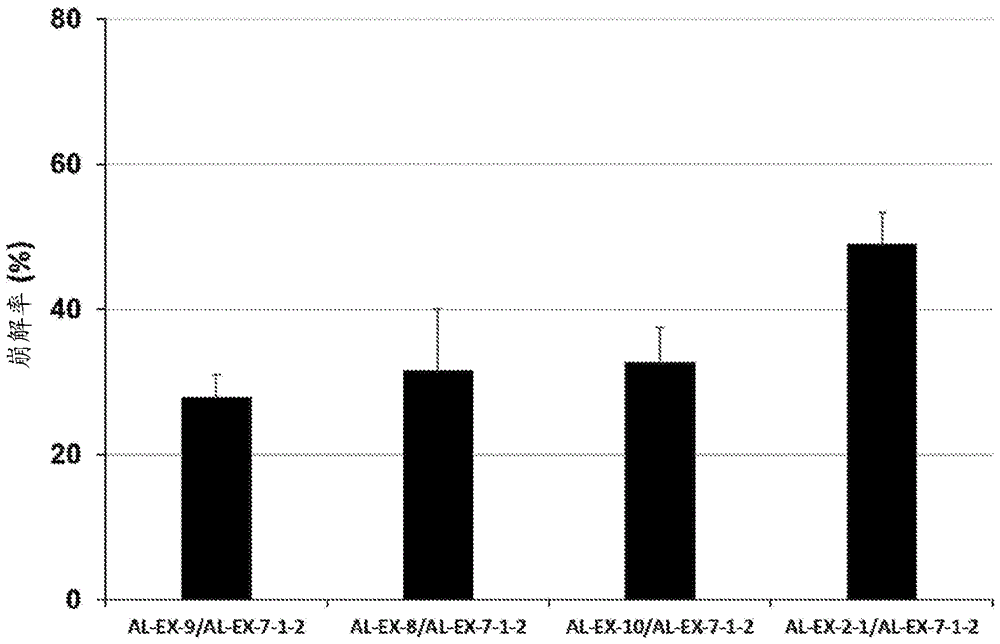
[图4]显示交联海藻酸结构体(al-ex-8/al-ex-7-1-2、al-ex-9/al-ex-7-1-2、al-ex-10/al-ex-7-1-2、或al-ex-2-1/al-ex-7-1-2)的edta处理后的凝胶稳定性的评价的图。
[图5]显示交联海藻酸结构体(al-ex-8/al-ex-7-1-2、al-ex-9/al-ex-7-1-2、al-ex-10/al-ex-7-1-2、或al-ex-2-1/al-ex-7-1-2)的凝胶透过率的评价的图。
具体实施方式
以下,对本发明详细进行说明。
1.海藻酸衍生物
在此,提供海藻酸衍生物。海藻酸衍生物为海藻酸的羧基的一部分通过连接基被交联基团(也称为“反应性基团”)取代而得。即,可举出例如海藻酸的任意1个以上的羧基和在两末端具有交联基团(z)和氨基的连接基(-l-)进行酰胺键合而得的物质(下述式al-1)(式中,z为交联基团;-l-为连接基。-l-可举出例如前述式(i)中的-a1-、或前述式(ii)中的-a2-等)。
[化25]
交联基团例如为丙烯酸残基或硫醇残基。丙烯酸残基或硫醇残基的两交联基团可通过迈克尔加成反应而容易地形成共价键。
作为丙烯酸残基,可举出例如可通过与硫醇残基的反应而形成迈克尔加成体的残基,具体而言,可举出丙烯酸、马来酸、马来酰亚胺、富马酸等。此外,作为硫醇残基,可举出例如可通过与丙烯酸残基的反应而形成迈克尔加成体的残基,具体而言,可举出hs-(ch2)m4-ph(m4=0~10,优选为m4=0~2)。作为硫醇残基,优选举出苯甲基硫醇、或硫基苯酚等。
前述交联基团可优选为可通过迈克尔加成反应而容易地形成迈克尔加成体的基团,例如,作为丙烯酸残基,为丙烯酰基,作为硫醇残基,为硫醇基;更优选为例如作为丙烯酸残基,为马来酰亚胺基,作为硫醇残基,为苯甲基硫醇基。
此外,交联基团可以键合有用于与交联基团和海藻酸二者键合而使二者保持一定距离的连接基(间隔物)。优选马来酰亚胺、或苯甲基硫醇或苯甲基硫醇的硫醇基保护体作为交联基团通过连接基而键合的海藻酸衍生物。
一些方式中,提供以下的海藻酸衍生物。
海藻酸衍生物,其中,在选自海藻酸、它的酯和它的盐中至少1种海藻酸类的羧基的一部分上具有下述式(i)所示的交联基团(反应性基团)(式中,虚线右侧除外):
[化26]
(式中,-a1-为选自下述式中的连接基(各式中,两虚线外侧除外):
[化27]
r1各自独立地为选自氢原子、甲基、异丙基、异丁基、仲丁基、羟基甲基、2-羟基乙基、硫醇甲基、甲基硫基乙基、羧基甲基、羧基乙基、氨基羰基甲基、氨基羰基乙基、氨基丁基、胍基丙基、苯甲基、4-羟基苯甲基、3-吲哚基甲基、4-咪唑基甲基和与r1所键合的碳原子和该碳原子所键合的氮原子一起形成环的丙烷-1,3-二基中的基团;
r2各自独立地为选自氢原子、甲基、异丙基、异丁基、仲丁基、羟基甲基、2-羟基乙基、硫醇甲基、甲基硫基乙基、羧基甲基、羧基乙基、氨基羰基甲基、氨基羰基乙基、氨基丁基、胍基丙基、苯甲基、4-羟基苯甲基、3-吲哚基甲基、4-咪唑基甲基和与r2所键合的碳原子和该碳原子所键合的氮原子一起形成环的丙烷-1,3-二基中的基团;
n为1~18的整数;
m为1~9的整数;
j为0~9的整数)。一些方式的海藻酸衍生物中,-a1-=-ch2ch2-的情况除外。
另一些方式中,提供以下的海藻酸衍生物。
海藻酸衍生物,其中,在选自海藻酸、它的酯和它的盐中至少1种海藻酸类的羧基的一部分上具有下述式(ii)所示的基团(式中,虚线右侧除外):
[化28]
(式中,p1为氢原子或硫醇基(-sh基)的保护基,
-a2-为下述式的连接基(式中,两虚线外侧除外):
[化29]
前述-a2-中,ar为可被水溶性取代基(例如,1~3个)取代的亚苯基;
n4为0~10的整数;
m4为0~10的整数;
p为0~10的整数)。
即,前述海藻酸衍生物更具体而言为
海藻酸类的任意1个以上的羧基与前述式(i)所示的交联基团形成酰胺键的下述式(al-1-i)所示的海藻酸衍生物:
[化30]
[式(al-1-i)中,连接基(-a1-)的定义如前所述];或者
海藻酸类的任意1个以上的羧基与前述式(ii)所示的交联基团形成酰胺键的下述式(al-1-ii)所示的海藻酸衍生物:
[化31]
[式(al-1-ii)中,p1和连接基(-a2-)的定义如前所述]。
在此,“具有式(i)的基团”、“被式(i)的基团取代”、“具有式(ii)的基团”或“被式(ii)的基团取代”是指选自海藻酸、它的酯和它的盐中至少1种中的羧基与式(i)的基团或式(ii)的基团(即,与交联基团键合的间隔物)的末端氨基形成酰胺键,由此选自海藻酸、它的酯和它的盐中至少1种与交联基团通过间隔物而键合。
式(i)和/或式(ii)的基团通过取代选自海藻酸、它的酯和它的盐中至少1种的海藻酸类(有时称为“海藻酸类”)的羧基的一部分而导入至海藻酸类。
式(al-1-i)或式(al-1-ii)所示的海藻酸衍生物的重均分子量优选为10万da~300万da,更优选为30万da~250万da,进一步优选为50万da~200万da。海藻酸衍生物的分子量可利用与前述海藻酸类相同的方法求出。
式(al-1-i)所示的海藻酸衍生物中,前述式(i)所示的交联基团、和式(al-1-ii)所示的海藻酸衍生物中,前述式(ii)所示的交联基团不需要分别与海藻酸类的构成单元的全部羧基键合。
式(al-1-i)所示的海藻酸衍生物和式(al-1-ii)所示的海藻酸衍生物中,各自的海藻酸衍生物中的式(i)的基团和式(ii)的基团的导入率(即,交联基团的导入率)各自优选为1%~30%,更优选为2%~15%,进一步优选为3%~10%。
式(al-1-i)所示的海藻酸衍生物和式(al-1-ii)所示的海藻酸衍生物中,各自的海藻酸衍生物中的式(i)的基团和式(ii)的基团的导入率(即,交联基团的导入率)分别是将在作为海藻酸类的重复单元的糖醛酸单糖单元中导入了交联基团的糖醛酸单糖单元的数值以百分数表示的值。只要没有特别限定,海藻酸衍生物(式(al-1-i)或式(al-1-ii))中的式(i)或式(ii)的基团的导入率中使用的%是指mol%。式(i)或式(ii)的基团的导入率可利用后述实施例所述的方法求出。
本说明书中,式(al-1-i)中的马来酰亚胺基和式(al-1-ii)中的硫醇基通过迈克尔加成反应而形成共价键(硫键),由此可形成交联。
1.1交联基团和连接基
在此,式(i)中,有时将部分结构式称为“交联基团”或“反应性基团”(式中虚线右侧除外):
[化32]
此外,有时将-a1-称为“间隔物”或“连接基”。
作为式(i)的间隔物(连接基)的-a1-选自下述式(各式中,两虚线外侧除外):
[化33]
(一些方式中,-a1-=-ch2ch2-的情况除外);
更优选选自下述式(各式中,虚线右侧除外):
[化34]
(一些方式中,-a1-=-ch2ch2-的情况除外)。
在此,下述式包含氨基酸或肽的部分结构:
[化35]
r1和r2如下所述为各氨基酸单元(-co-(chr)-nh-)或(-nh-(chr)-co-)的侧链(r)。
(1)氢原子(甘氨酸的侧链)
(2)甲基(丙氨酸的侧链)
(3)异丙基(缬氨酸的侧链)
(4)异丁基(亮氨酸的侧链)
(5)仲丁基(异亮氨酸的侧链)
(6)羟基甲基(丝氨酸的侧链)
(7)2-羟基乙基(苏氨酸的侧链)
(8)硫醇甲基(半胱氨酸的侧链)
(9)甲基硫基乙基(蛋氨酸的侧链)
(10)羧基甲基(天冬氨酸的侧链)
(11)羧基乙基(谷氨酸的侧链)
(12)氨基羰基甲基(天冬酰胺的侧链)
(13)氨基羰基乙基(谷氨酰胺的侧链)
(14)氨基丁基(赖氨酸的侧链)
(15)胍基丙基(精氨酸的侧链)
(16)苯甲基(苯丙氨酸的侧链)
(17)4-羟基苯甲基(酪氨酸的侧链)
(18)3-吲哚基甲基(色氨酸的侧链)
(19)4-咪唑基甲基(组氨酸的侧链)
(20)与r1所键合的碳原子和该碳原子所键合的氮原子一起形成环的丙烷-1,3-二基(脯氨酸的侧链)
前述各方式中,n优选为1~10的整数,更优选为1~8的整数,进一步优选为3~6的整数。
前述各方式中,m优选为1~7的整数,更优选为1~5的整数,进一步优选为1~3的整数,特别优选为1或2。
前述各方式中,j优选为0~8的整数,更优选为1~6的整数,进一步优选为2~4的整数,特别优选为0或1。
本说明书中,式(i)中的连接基-a1-所含的下述式(al-a1-1)或式(al-a1-2)中(两式中,虚线两外侧不包括在内):
[化36]
式中存在不对称碳时,表示也包含其各光学异构体。
例如,表示式(i)中的连接基-a1-为下述式时(al-a1-1-a)(式中,虚线两外侧不包括在内):
[化37]
包括苯甲基取代的碳的立体构型为s体的下述式(al-a1-1-as)和苯甲基取代的碳的立体配置为r体的下述式(al-a1-1-ar)所示的连接基(所有式中,虚线两外侧不包括在内):
[化38]
此外,例如,表示作为式(i)之一的式(i-x)(式中,虚线右侧不包括在内)中:
[化39]
存在光学异构体,只要没有特别限定,则包含s体(式(i-x-s))、或r体(式(i-x-r))所示的异构体。
[化40]
本发明的式(i)中,连接基-a1-中存在不对称碳时(为光学活性体时),在合成与式(i)对应的胺衍生物(am-i)的步骤中,利用通常的光学拆分手段(分离手法)能由其外消旋体分离为各光学活性体,此外,在合成与式(i)对应的胺衍生物(am-i)的步骤中,通过使用不对称合成,可选择性地合成光学异构体的一者,能合成各光学活性体。通过使用所得各光学活性的胺衍生物,能合成可导入具有不对称碳的(光学活性的)式(i)的基团的海藻酸衍生物。
作为前述分离方法,可举出例如分级重结晶法、非对映体法、和手性柱法等光学拆分法。以下对各拆分法进行详述。
分级重结晶法:使外消旋体与光学拆分剂发生离子键合,得到结晶性的非对映体后,通过分级重结晶法分离该结晶性的非对映体,根据期望经过光学拆分剂的除去步骤,得到光学上纯粹的化合物的方法。光学拆分剂可举出例如(+)-扁桃酸、(-)-扁桃酸、(+)-酒石酸、(-)-酒石酸、(+)-1-苯乙基胺、(-)-1-苯乙基胺、金鸡纳宁、(-)-金鸡纳定、和马钱子碱等。
非对映体法:使外消旋体的混合物与光学拆分剂发生共价键合,得到非对映体的混合物,接着,通过通常的分离手段(例如,分级重结晶、硅胶柱色谱、和hplc等)分离为光学上纯粹的非对映体,然后,经过基于化学反应(水解反应等)的光学拆分剂的除去步骤,得到光学上纯粹的光学异构体的反应。
例如,本发明的化合物或中间体化合物具有羟基或氨基(伯、仲)时,通过该化合物与光学活性有机酸(例如,α-甲氧基-α-(三氟甲基)苯基乙酸、和(-)-薄荷氧基乙酸等)的缩合反应,分别得到酯体或酰胺体的非对映体。此外,本发明的化合物具有羧基时,通过该化合物与光学活性胺或光学活性醇的缩合反应,分别得到酰胺体或酯体的非对映体。分离通过缩合反应得到的非对映体,通过将各非对映体供于基于酸或碱的水解反应,转化为原化合物的光学上纯粹的光学异构体。
手性柱法:通过将外消旋体或它的盐供于基于手性柱(光学异构体分离用柱)的色谱从而直接进行光学拆分的方法。
例如,高效液相色谱(hplc)时,在手性柱(例如,ダイセル公司制chiralシリーズ等)中添加光学异构体的混合物,使用溶出溶剂(水、各种缓冲液(例如磷酸缓冲液)、和有机溶剂(例如乙醇、甲醇、异丙醇、乙腈、三氟乙酸、和二乙基胺等)等单独溶剂、或它们的混合溶剂)展开,由此能分离光学异构体。此外,例如,气体色谱时,使用手性柱(例如,cp-chirasil-dexcb(ジーエルサイエンス公司制)等),能分离光学异构体。此外,例如,超临界流体色谱(sfc)时,在手性柱(例如,ダイセル公司制chiralシリーズ等)中添加光学异构体的混合物,在溶出溶剂中使用二氧化碳和适当的有机溶剂(例如甲醇、乙醇、异丙醇、三氟乙酸、和二乙基胺等),能分离光学异构体。
作为选择性合成前述光学异构体一者的不对称合成,可举出(1)使外消旋化合物对映选择性地反应而导入学活性体的不对称合成反应、(2)从天然存在的光学活性化合物(糖、氨基酸等)选择性地合成非对映体的方法等。
前述各方式中,-a1-进一步优选选自下述式(各式中,两虚线外侧除外):
[化41]
(一些方式中,-a1-=-ch2ch2-的情况除外)。
前述各方式中,-a1-特别优选选自下述式(各式中,两虚线外侧除外):
[化42]
(一些方式中,-a1-=-ch2ch2-的情况除外)。
更优选为式(i)所示的基团选自下述式(各式中,虚线右外侧除外):
[化43]
(一些方式中,下述式的基团除外(各式中,虚线右外侧除外)):
[化44]
进一步优选为式(i)所示的基团选自下述式(各式中,虚线右外侧除外):
[化45]
(一些方式中,下述式的基团除外(各式中,虚线右外侧除外)):
[化46]
此外,前述式(ii)中,有时将部分结构称为“交联基团”或“反应性基团”:
[化47]
此外,有时将-a2-称为“间隔物”或“连接基”。
式(ii)的交联基团中p1为氢原子或硫醇基(-sh基)的保护基。作为该保护基,可举出例如乙酰基、苯甲酰基、三苯基甲基、甲氧基甲基、和n-乙基氨基甲酸酯基等保护基,优选为乙酰基、和苯甲酰基,更优选为乙酰基。
p1优选为氢原子、乙酰基或苯甲酰基,更优选为氢原子、或乙酰基。
作为式(ii)的连接基的-a2-为下述式所示的连接基(式中,两虚线外侧除外):
[化48]
前述各方式中,n4优选为0~8的整数,更优选为0~6的整数,进一步优选为0~2的整数,特别优选为0或2。
前述各方式中,m4优选为0~8的整数,更优选为0~6的整数,进一步优选为0~2的整数,特别优选为1。
前述各方式中,p优选为0~8的整数,更优选为1~6的整数,进一步优选为2~4的整数,特别优选为2或3。
前述各方式中,ar为可被水溶性取代基取代的亚苯基,例如,邻亚苯基、间亚苯基、或对亚苯基,优选为对亚苯基。这些方式中,亚苯基可独立地被例如1~4个、优选为1~3个、更优选为1~2个的水溶性取代基取代。
本说明书中,亚苯基是指可从苯环上去除2个氢的多价基团,表示为-c6h4-。亚苯基可举出例如去除互为邻位的2个氢原子而得的邻亚苯基(邻亚苯基)、除去互为间位的2个氢原子而得的间亚苯基(间亚苯基)、除去互为对位的2个氢原子而得的对亚苯基(对亚苯基)。
本说明书中,水溶性取代基例如可举出羟基(-oh)、羧基(-cooh)、氨基(-nh2)、硫醇基(-sh)或磺基(-so2oh)等取代基,优选举出羟基和氨基。
前述各方式中,ar为未取代的亚苯基(例如,邻亚苯基、间亚苯基或对亚苯基,优选为对亚苯基)。
前述各方式中,-a2-进一步优选为选自下述式中的连接基(各式中,两虚线外侧除外):
[化49]
-ar-为对亚苯基。
更优选为式(ii)所示的基团选自下述式(各式中,虚线右侧除外):
[化50]
进一步优选为式(ii)所示的基团选自下述式(各式中,虚线右侧除外):
[化51]
一些方式中,式(i)和/或式(ii)的交联基团(反应性基团)为通过迈克尔反应形成迈克尔加成体而由此进行交联反应的基团即可。此外,通过导入间隔物(连接基),即使在交联基团的导入率低的情况下交联反应也进行。通过交联反应,海藻酸衍生物通过交联而形成三维网眼结构。优选海藻酸衍生物的交联后的稳定性与交联前相比得到改善的海藻酸衍生物。
1.2海藻酸类
所使用的海藻酸类可以源自天然,也可以为合成物,优选源自天然。优选使用的海藻酸类为由巨藻、巨囊藻、海带、泡叶藻、丛梗藻、褐藻苷苔、黑海带、昆布等褐藻类提取的生物体内吸收性的多糖类,是d-甘露糖醛酸(m)和l-葡糖醛酸(g)这2种糖醛酸聚合为直链状的聚合物。更具体而言,是d-甘露糖醛酸的均聚物组分(mm组分)、l-葡糖醛酸的均聚物组分(gg组分)、和d-甘露糖醛酸与l-葡糖醛酸无规排列而得的组分(m/g组分)任意键合而得的嵌段共聚物。本说明书中,记载为海藻酸类时,是指选自海藻酸、海藻酸酯、和它们的盐(例如,海藻酸钠)中的至少1种海藻酸。
本说明书中,海藻酸、海藻酸衍生物、交联海藻酸、和交联海藻酸的分子量中,作为单位,有时计作da(道尔顿)。
海藻酸类的d-甘露糖醛酸和l-葡糖醛酸的构成比(m/g比)主要根据源自海藻等的生物的种类而不同,此外,受到由该生物的生育场所、季节带来的影响,遍及m/g比为约0.2的高g型至m/g比为约5的高m型的高范围。海藻酸类的凝胶化能力和所生成的凝胶的性质根据m/g比而受到影响,一般已知g比率高时凝胶强度变高。除此之外,m/g比对凝胶的硬度、脆度、吸水性、柔软性等也带来影响。所使用的海藻酸类和/或其盐的m/g比通常为0.2~4.0,更优选为0.4~3.0、进一步优选为0.5~3.0。
本说明书中,使用“~”表示的数值范围表示包含“~”前后记载的数值作为各自最小值和最大值的范围。
所使用的“海藻酸酯”、“海藻酸盐”没有特别限定,为了与交联剂反应,需要不具有阻碍交联反应的官能团。作为海藻酸酯,优选举出海藻酸丙二醇酯。
作为海藻酸盐,可举出例如海藻酸的1价盐、海藻酸的2价盐。
作为海藻酸的1价盐,优选举出海藻酸钠、海藻酸钾、海藻酸铵等,更优选为海藻酸钠或海藻酸钾,尤其优选为海藻酸钠。
作为海藻酸的2价盐,优选举出海藻酸钙、海藻酸镁、海藻酸钡、海藻酸锶等。
海藻酸类为高分子多糖类,难以正确地确定分子量,一般为重均分子量为1000~1000万、优选为1万~800万、更优选为2万~300万的范围。源自天然物的高分子物质的分子量测定中,已知根据测定方法不同会产生值的差异。
例如,通过凝胶浸透色谱(gpc)或凝胶过滤色谱(它们也被统称为尺寸排除色谱)测得的重均分子量优选为10万以上、更优选为50万以上,且优选为500万以下、更优选为300万以下。其优选范围为10万~500万,更优选为15万~300万。
此外,例如,根据gpc-mals法,可以测定绝对重均分子量。通过gpc-mals法测得的重均分子量(绝对分子量)优选为1万以上、更优选为5万以上、进一步优选为6万以上,且优选为100万以下、更优选为80万以下、进一步优选为70万以下、特别优选为50万以下。其优选范围为1万~100万,更优选为5万~80万,进一步优选为6万~70万、特别优选为6万~50万。
通常利用上述方法计算高分子多糖类的分子量时,会产生10%~20%的测定误差。例如,若为40万,则在32万~48万左右的范围产生值的浮动,若为50万,则在40万~60万左右的范围产生值的浮动,若为100万,则在80万~120万左右的范围产生值的浮动。
海藻酸类的分子量的测定可根据常规方法测定。
分子量测定中使用凝胶过滤色谱时的代表的条件如后述本说明书的实施例所述。柱例如可使用superose6increase10/300gl柱(ge医疗科学公司),作为展开溶剂,例如可使用包含0.15mol/lnacl的10mmol/l磷酸缓冲液(ph7.4),作为分子量标准,可以使用蓝色葡聚糖、甲状腺球蛋白、铁蛋白、醛缩酶、伴白蛋白、卵白蛋白、核糖核酸酶a和抑肽酶。
所使用的海藻酸类的粘度没有特别限定,测定粘度作为1w/w%的海藻酸类的水溶液时,优选为10mpa・s~1000mpa・s、更优选为50mpa・s~800mpa・s。
海藻酸类的水溶液的粘度的测定可以根据常规方法测定。例如,可以使用旋转粘度计法的同轴双筒型形旋转粘度计、单筒型旋转粘度计brookfield型粘度计)、圆锥-平板型旋转粘度计(锥板型粘度计)等测定。优选期望根据日本药典(第16版)的粘度测定法。更优选使用锥板型粘度计。
海藻酸类由褐藻类提取而得,最初分子量大、粘度高,但通过基于热的干燥、纯化等过程中,分子量变小,粘度变低。通过制造步骤的温度等条件管理、作为原料的褐藻类的选择、制造步骤中的分子量的分级等方法可制造分子量不同的海藻酸类。进一步,通过与具有不同分子量或者粘度的其他种类的海藻酸类混合,也能形成具有目标分子量的海藻酸类。
所使用的海藻酸类在一些方式中不是低内毒素海藻酸。在另一些方式中,海藻酸类为低内毒素海藻酸。低内毒素是指内毒素水平低至不实质性引发炎症、或发热的程度。更优选期望为经低内毒素处理的海藻酸类。
低内毒素处理可以通过公知的方法或基于其的方法进行。例如,可以通过纯化透明质酸钠的菅等的方法(例如,参照日本特开平9-324001号公报等)、纯化β1,3-葡聚糖的吉田等的方法(例如,参照日本特开平8-269102号公报等)、纯化海藻酸钠、结冷胶等生物体高分子盐的威廉等的方法(例如,参照特表2002-530440号公报等)、纯化多糖的詹姆斯等的方法(例如,参照国际公开第93/13136号单行本等)、路易斯等的方法(例如,参照美国特許第5589591号说明书等)、纯化海藻酸盐的赫尔曼弗兰克等的方法(例如,参照applmicrobiolbiotechnol(1994)40:638-643等)等或基于它们的的方法而实施。低内毒素处理不限于它们,可以通过洗涤、基于过滤器(除去内毒素的过滤器、帯电过滤器等)的过滤、超滤、使用柱(内毒素吸附亲和柱、凝胶过滤柱、基于离子交换树脂的柱等)的纯化、在疏水性物质、树脂或活性炭等中的吸附、有机溶剂处理(基于有机溶剂的提取、添加有机溶剂导致的的析出・沉降等)、表面活性剂处理(例如,参照日本特开2005-036036号公报等)等公知的方法、或者适当组合它们而实施。在这些处理的步骤中可以适当组合离心分离等公知的方法。期望根据海藻酸的种类而适当选择。
内毒素水平可利用公知的方法确认,例如,可以通过利用鲎试剂(lal)的方法、使用エントスペシー(注册商标)es-24sset(生化学工业株式会公司)的方法等测定。
所使用的内毒素的处理方法没有特别限定,作为其结果,进行利用鲎试剂(lal)的内毒素测定时,海藻酸类的内毒素含量优选为500内毒素单位(eu)/g以下,进一步优选为100eu/g以下,尤其优选为50eu/g以下,特别优选为30eu/g以下。经低内毒素处理的海藻酸钠可通过例如seamatrix(注册商标)(持田制药株式会公司)、pronovatmuplvg(fmcbiopolymer)等市售品而获得。
1.3组合物
在此,提供组合物,其包含海藻酸类的任意1个以上的羧基与前述式(i)所示的交联基团形成酰胺键的下述式(al-1-i)所示的至少1种的海藻酸衍生物、和海藻酸类的任意1个以上的羧基与前述式(ii)所示的交联基团形成酰胺键的下述式(al-1-ii)所示的至少1种的海藻酸衍生物。应予说明,式(al-1-i)的连接基(-a1-)的各方式、和式(al-1-ii)的连接基(-a2-)和p1的各方式如前所述。
[化52]
一些方式的组合物中,式(al-1-i)的海藻酸衍生物与式(al-1-ii)的海藻酸衍生物的重量比(式(al-1-i)的海藻酸衍生物:式(al-1-ii)的海藻酸衍生物)例如为1:1~1.5、优选为1:1.2~1.5或1:1~1.2、更优选为1:1。
一些方式的组合物中,式(al-1-ii)的海藻酸衍生物与式(al-1-i)的海藻酸衍生物的重量比(式(al-1-ii)的海藻酸衍生物:式(al-1-i)的海藻酸衍生物)例如为1:1~1.5、优选为1:1.2~1.5或1:1~1.2、更优选为1:1。
一些方式的组合物中,式(al-1-i)的海藻酸衍生物与式(al-1-ii)的海藻酸衍生物的混合比以式(al-1-i)的海藻酸衍生物与式(al-1-ii)的海藻酸衍生物的交联基团(反应性基团)的导入率(mol%)比计,例如为1:1~1.5、优选为1:1.2~1.5或1:1~1.2、更优选为1:1。
一些方式的组合物中,式(al-1-ii)的海藻酸衍生物与式(al-1-i)的海藻酸衍生物的混合比以式(al-1-ii)的海藻酸衍生物与式(al-1-i)的海藻酸衍生物的交联基团(反应性基团)的导入率(mol%)比计,例如为1:1~1.5、优选为1:1.2~1.5或1:1~1.2、更优选为1:1。
1.4交联海藻酸结构体
交联海藻酸结构体为式(al-1-i)的海藻酸衍生物、式(al-1-ii)的海藻酸衍生物、或它们的混合物(以下简记为“海藻酸衍生物”)通过交联基团形成三维网眼结构的结构体。交联海藻酸结构体可通过对具有交联基团的海藻酸衍生物实施交联反应而得到。交联反应例如如下所述或者可将它们适当组合而实施,并不限定于它们。
(a)基于使包含式(al-1-i)的海藻酸衍生物的组合物与包含式(al-1-ii)的海藻酸衍生物的组合物反应的交联反应(共价键交联反应)、或
(b)基于使包含式(al-1-i)的海藻酸衍生物或式(al-1-ii)的海藻酸衍生物的组合物在包含2价金属离子(例如,钙离子、钡离子等)的溶液中反应的交联反应(离子键交联反应)。
(c)基于使包含式(al-1-i)的海藻酸衍生物和式(al-1-ii)的海藻酸衍生物的组合物在包含2价金属离子(例如,钙离子、钡离子等)的溶液中反应的交联反应(共价键交联反应+离子键交联反应)。
关于交联海藻酸结构体的制备方法,在后述2.2中记载。
交联海藻酸结构体的形状没有特别限定,例如,可举出管状结构体、纤维状结构体、纤维、珠、凝胶、近球形的凝胶、微囊等,优选为纤维、珠或近球形的凝胶。
优选交联海藻酸结构体的稳定性得到改善。此外,交联海藻酸结构体可具有在其内部保持内容物的能力(内容物保持性)。
交联海藻酸结构体的稳定性可通过例如测定凝胶稳定性、测定凝胶泄漏率等而确认。
凝胶稳定性可如以下方式求出。在装入容器的交联海藻酸结构体凝胶中添加磷酸缓冲生理食盐水(pbs),测定在pbs中溶出的海藻酸的浓度(μg/ml)。将测定得到的海藻酸浓度除以通过分解交联海藻酸结构体凝胶而得的总海藻酸浓度,将得到的值以百分数表示的值作为崩解率。凝胶稳定性具体而言可利用后述实施例所述的方法求出。交联海藻酸结构体的凝胶崩解率优选为0%~90%,更优选为0%~70%,进一步优选为0%~50%。在水溶液中溶出的海藻酸的浓度越低,即凝胶崩解率越低则交联海藻酸结构体的稳定性越提高。
凝胶泄漏率可如以下方式求出。制作内包异硫氰酸荧光素-葡聚糖的交联海藻酸结构体凝胶,在装入容器的前述凝胶中添加磷酸缓冲生理食盐水(pbs),测定在pbs中泄漏的葡聚糖浓度。将测定的葡聚糖的浓度除以通过分解内包异硫氰酸荧光素-葡聚糖的交联海藻酸结构体凝胶而得的总葡聚糖浓度得到的值以百分数表示的值为凝胶泄漏率。凝胶泄漏率具体而言可利用后述实施例所述的方法求出。交联海藻酸衍生物的添加pbs48小时后的凝胶泄漏率优选为0%~90%,更优选为0%~70%,进一步优选为0%~50%。凝胶泄漏率越低则交联海藻酸衍生物的稳定性越提高。
在此,内包异硫氰酸荧光素-葡聚糖的交联海藻酸结构体凝胶为如下制备的凝胶。混合具有交联基团的海藻酸衍生物的溶液和异硫氰酸荧光素-葡聚糖溶液,将该混合溶液滴加在包含钙离子的溶液中,将所得凝胶在溶液中在37℃下静置10分钟引起交联反应,由此可得到内包异硫氰酸荧光素-葡聚糖的交联海藻酸结构体凝胶。
2.海藻酸衍生物的合成方法
海藻酸衍生物可通过使导入了交联基团(反应性基团)的连接基末端氨基和海藻酸类的羧基发生缩合反应而得到。
具体而言,式(al-1-i)或式(al-1-ii)所示的海藻酸衍生物可以分别将式(am-i)所示的胺衍生物(式中,-a1-与前述一些方式中的定义相同)、或式(am-ii)所示的胺衍生物(式中,p1和-a2-与前述一些方式中的定义相同)与海藻酸类的任意羧基使用缩合剂进行缩合反应而制造。
[化53]
[式(al-1-i)的海藻酸衍生物的制备方法]
使用0.5重量%~1重量%的海藻酸水溶液和式(am-i)所示的氨基衍生物,按照文献公知的方法、例如“实验化学讲座第5版16、有机化合物的合成iv、羧酸和衍生物、酯类、p35-70、酸酰胺和酸酰亚胺、p118-154、氨基酸・肽、p258-283、2007年、丸善”等中记载的方法,在选自1,3-二环己基碳二酰亚胺(dcc)、1-乙基-3-(3-二甲基氨基丙基)碳二酰亚胺盐酸盐(wsc・hcl)、苯并三唑-1-基氧基三(二甲基氨基)鏻六氟磷酸盐(bop试剂)、双(2-氧代-3-噁唑烷基)次膦酰氯(bop-cl)、2-氯-1,3-二甲基咪唑啉鎓六氟磷酸盐(cip)、或4-(4,6-二甲氧基-1,3,5-三嗪-2-基)-4-甲基吗啉鎓氯化物(dmt-mm)等中的缩合剂的存在下、在海藻酸不析出程度的选自四氢呋喃、1、4-二氧杂环己烷等醚系溶剂、甲醇、乙醇、2-丙醇等醇系溶剂、n,n-二甲基甲酰胺等极性溶剂等中的溶剂和水的混合溶剂中,在碳酸氢钠、碳酸钠等无机碱、或三乙基胺、吡啶等有机碱的存在下或非存在下,在0℃至50℃间的温度下进行缩合反应,由此可制造式(al-1-i)的海藻酸衍生物。
[式(al-1-ii)的海藻酸衍生物的制备方法]
使用0.5重量%~1重量%的海藻酸水溶液和式(am-ii)所示的氨基衍生物,按照前述[式(al-1-i)的海藻酸衍生物的制备方法]进行反应,由此可制造式(al-1-ii)的海藻酸衍生物。
前述式(al-1-i)的海藻酸衍生物或式(al-1-ii)的海藻酸衍生物的制备方法中,式(am-i)或式(am-ii)的氨基衍生物的导入率由于考虑该氨基衍生物的性质等,可通过适当选择下述(i)~(v)等反应条件并组合而调节。为了(i)缩合剂的等量增减、(ii)反应温度的上升・下降、(iii)反应时间的延长・缩短、(iv)反应基质的海藻酸的浓度的调整、(v)提高式(am-1)或式(am-2)的氨基衍生物的溶解度,添加与水混合的有机溶剂等。
式(al-1-ii)的p1为硫醇保护体(乙酰基、苯甲酰基等)时,使用相对于导入的保护硫醇基为过量的氢氧化钠、氢氧化钾等无机碱使0.5重量%~1重量%的该硫醇保护海藻酸衍生物的水溶液在0℃至30℃下水解,由此可制造导入硫醇基的海藻酸衍生物(式(al-1-ii)中p1为氢原子),中和过剩的碱,可直接以溶液形式用于交联反应。
以下示出式(am-i)或式(am-ii)所示的氨基衍生物的制造方法。
2.1氨基衍生物的合成
2.1.1式(am-i)的氨基衍生物的合成
(反应式a)
[化54]
(反应式a)<步骤1>
使用式(iii)[式(iii)的化合物为市售化合物或可由市售化合物利用文献公知的制造方法制造的化合物。式中p2为氨基的保护基,可适当选择]所示的胺,按照文献公知的方法、例如(实验化学讲座第5版16、有机化合物的合成iv、羧酸和衍生物、酸酰胺和酸酰亚胺、146-154页、2007年、丸善)等中记载的方法,与马来酸在1,3-二环己基碳二酰亚胺(dcc)、1-乙基-3-(3’-二甲基氨基丙基)碳二酰亚胺盐酸盐(wsc・hcl)、苯并三唑-1-基氧基三(二甲基氨基)鏻六氟磷酸盐(bop试剂)、双(2-氧代-3-噁唑烷基)次膦酰氯(bop-cl)、2-氯-1,3-二甲基咪唑啉鎓六氟磷酸盐(cip)、4-(4,6-二甲氧基-1,3,5-三嗪-2-基)-4-甲基吗啉鎓氯化物(dmt-mm)等缩合剂的存在下,在1,4-二氧杂环己烷等醚系溶剂、二氯甲烷等卤素系溶剂、n,n-二甲基甲酰胺等极性溶剂等溶剂中,在碳酸氢钠、碳酸钠等无机碱、三乙基胺、吡啶等有机碱的存在下或非存在下,在0℃至50℃的温度下反应,由此可制造式(iv)的化合物。
此外,使马来酸酐与式(iii)所示的胺在甲醇、乙醇等醇系溶剂等溶剂中在三乙基胺等碱存在下或非存在下反应,由此也可制造式(iv)的化合物。
(反应式a)<步骤2>
使式(iv)所示的单酰胺在1,4-二氧杂环己烷等醚系溶剂、甲苯等烃系溶剂、1,2-二氯乙烷等卤素系溶剂等不影响反应的溶剂中,或者在乙酸酐溶剂中使用乙酸钠等碱在40℃至溶剂回流的温度(例如,100℃)下加热,由此可制造式(vi)的化合物,在<步骤1>的操作实施后不进行单酰胺体分离,在乙酸酐中使用乙酸钠等碱进行环化处理,由此也可制造式(vi)的化合物。
此外,使用适当的缩合剂,导入至活性酯,由此也可制造式(vi)的化合物。
(反应式a)<步骤3>
将式(v)[式(v)的化合物为市售化合物或可由市售化合物利用文献公知的制造方法制造的化合物]所示的醇和马来酰亚胺(1h-吡咯-2,5-二酮)在三苯基膦等膦试剂存在下,在四氢呋喃等醚系溶剂、甲苯等烃系溶剂等溶剂中,将偶氮二羧酸二乙基酯、偶氮二羧酸二异丙基酯等光延试剂在-78℃至室温左右的温度下反应,由此也可制造式(vi)的化合物。
(反应式a)<步骤4>
将式(vi)所示的保护体按照文献公知的方法例如(实验化学讲座第5版16、有机化合物的合成iv、氨基酸・肽、258-283页、2007年、丸善)等中记载的方法进行脱保护、并按照文献公知的方法例如“有机合成中的保护基团(protectivegroupsinorganicsynthesis4thedition)第4版、2007年、ジョンウィリーアンドサンズ(johnwiley&sons)、グリーン(greene)等”书本中记载的脱保护的方法进行脱保护,由此可制造式(am-i)的化合物。
应予说明,式(am-i)的胺衍生物的制备方法中的p2为选自-c(o)o-t-bu基、-c(o)o-bn基、-c(o)ch3基、-c(o)cf3基、-so2ph、-so2phme基、-so2ph(no2)基等中的氨基的保护基。
例如,p1为叔丁氧基羰基(-c(o)o-t-bu基)时,通过使用氯化氢、三氟乙酸等酸可进行脱保护。此外,可使用包含氯化氢的1,4-二氧杂环己烷、环戊基甲基醚、乙酸乙酯等。此外,三氟乙酸也可使用无溶剂或对酸非活性的二氯甲烷、甲苯等溶剂。
式(am-i)的胺根据需要可以盐酸盐、三氟乙酸盐等盐的形式得到。
2.1.2式(am-ii)的胺衍生物的合成
(反应式b)
[化55]
(反应式b)<步骤1>
使用式(viii)[式(viii)的化合物为市售化合物或可由市售化合物利用文献公知的制造方法制造的化合物。式中p2为氨基的保护基,可适当选择]所示的硫醇体,按照文献公知的方法例如(有机合成中的保护基团第3版protectionforthethiolgroup、457-486页、1999年)等中记载的方法,将乙酰基氯、苯甲酰基氯等酰氯、或三苯基甲基氯化物等烷基卤化物、或乙基异氰酸酯等异氰酸酯等在三乙基胺、吡啶等有机碱、碳酸氢钾等无机碱存在下或非存在下,使1,4-二氧杂环己烷等醚系溶剂、二氯甲烷等卤素系溶剂等对反应非活性的溶剂中反应,或者与羧酸衍生物在适当的缩合剂、酸催化剂中反应,由此可制造式(ix)的硫醇保护体。
(反应式b)<步骤2>
使用式(xi)[式(xi)的化合物为市售化合物或可由市售化合物利用文献公知的制造方法制造的化合物。式中p2为氨基的保护基,可适当选择]所示的卤素取代体(x=cl、br、i),将硫代苯甲酸、硫代乙酸、硫代乙酸钾等酰基硫代衍生物等在乙腈、二氯甲烷、n、n-二甲基甲酰胺等对反应非活性的溶剂中在碳酸钾等碱存在下或非存在下反应,由此可制造式(ix)的化合物。
(反应式b)<步骤3>
将式(ix)所示的n-保护体按照文献公知的方法例如(实验化学讲座第5版16、有机化合物的合成iv、氨基酸・肽、258-283页、2007年、丸善)等中记载的方法进行脱保护、并按照文献公知的方法例如“有机合成中的保护基团(protectivegroupsinorganicsynthesis4thedition)第4版、2007年、ジョンウィリーアンドサンズ(johnwiley&sons)、グリーン(greene)等”书本中记载的脱保护的方法进行脱保护,由此可制造式(am-ii)的化合物。
应予说明,式(am-ii)的胺衍生物的制备方法中的p2为选自-c(o)o-t-bu基、-c(o)o-bn基、-c(o)ch3基、-c(o)cf3基、-so2ph、-so2phme基、-so2ph(no2)基等中的氨基的保护基。
例如,p1为叔丁氧基羰基(-c(o)o-t-bu基)时,通过使用氯化氢、三氟乙酸等酸可进行脱保护。此外,可使用包含氯化氢的1,4-二氧杂环己烷、环戊基甲基醚、乙酸乙酯等。此外,三氟乙酸也可使用无溶剂或对酸非活性的二氯甲烷、甲苯等溶剂。
式(am-ii)的胺根据需要可以盐酸盐、三氟乙酸盐等盐的形式得到。
2.2交联海藻酸结构体的制备
交联海藻酸结构体可通过包括对具有交联基团的海藻酸衍生物实施前述交联反应的方法而得到。具体而言,例如可通过以下的方法制备,但不限定于它们。
(a)涂布法
将包含前述式(al-1-i)的海藻酸衍生物的溶液滴加在包含2价金属离子的溶液中等部分地进行交联,得到特定的结构体。通过将前述得到的例如凝胶等结构体添加在包含前述式(al-1-ii)的海藻酸衍生物的溶液中,对前述结构体的表面等进一步实施交联反应,由此可得到交联海藻酸结构体。应予说明,该方法也可分别地将式(al-1-i)的海藻酸衍生物置换为式(al-1-ii)的海藻酸衍生物而实施,将式(al-1-ii)的海藻酸衍生物置换为式(al-1-i)的海藻酸衍生物而实施。
(b)混合法
混合包含前述式(al-1-i)的海藻酸衍生物的溶液和包含前述式(al-1-ii)的海藻酸衍生物的溶液,将该混合溶液滴加在包含2价金属离子的溶液中等部分地进行交联,得到为特定的结构体的交联海藻酸结构体。
作为上述方法中使用的2价金属离子,具体而言,可举出例如钙离子、镁离子、钡离子、锶离子、锌离子等,优选为钙离子。包含前述钙离子的溶液的钙离子浓度没有特别限定,可举出例如1mm~1m,优选为5mm~500mm,更优选为10mm~300mm。
此外,交联反应时使用的溶剂或溶液也没有特别限定,可举出例如超纯水、细胞培养用培养基、磷酸缓冲生理食盐水(pbs)、和生理食盐水,优选为超纯水。作为前述特定的结构体,可举出例如管状结构体、纤维状结构体、纤维、珠、凝胶、近球形的凝胶、微囊等。
3海藻酸衍生物、交联海藻酸结构体的用途
海藻酸衍生物在食品、医疗、化状品、纤维、制纸等广泛的领域中可代替以往的海藻酸使用。作为海藻酸衍生物或交联海藻酸结构体的优选用途,具体而言,可举出创伤包覆材料、术后防粘连材料、药剂缓释用基材、细胞培养用基材、细胞移植用基材等医疗用材料。
作为用作医疗用材料时的交联海藻酸结构体的形状,可举出管状、纤维状、纤维、珠、凝胶、近球形的凝胶等,优选为珠、凝胶或近球形的凝胶,更优选为近球形的凝胶。
应予说明,本说明书中引用的全部发行物、例如现有技术文献、和公开公报、专利公报和其他专利文献作为参照组合入本说明书中。本说明书包含作为本申请的优先权基础的日本专利申请即日本特愿2018-062201号(2018年3月28日申请)的权利要求书、说明书、和附图的公开内容。
并且,本发明的目的、特征、优点及其构思通过本说明书的记载,对于本领域技术人员来说是显而易见的,根据本说明书的记载,只要是本领域技术人员,就能够容易地实施本发明。用于实施发明的最佳方式以及具体的实施例等表示本发明的优选实施方式,是为了例示或者说明而示出的,本发明不限于它们。对于本领域技术人员来说显而易见的是,在本说明书公开的本发明的意图和范围内,基于本说明书的记载,可以进行各种修改。
实施例
核磁共振光谱(nmr)的测定使用jeoljnm-ecx400ft-nmr(日本电子)。
1h-nmr数据中,nmr信号模式中,s表示单重态,d表示双重态,t表示三重态,q表示四重态,m表示多重态,br表示宽范围,j表示偶联常数,hz表示赫兹、cdcl3表示氘代氯仿、dmso-d6表示氘代二甲基亚砜、d2o表示氘代水。1h-nmr数据中,关于羟基(oh)、氨基(nh2)、羧基(cooh)的质子等由于为宽带而无法确认的信号,未记载数据。
实施例中的“室温”通常表示约0℃至约35℃的温度。
实施例中的“导入率”是在d2o中进行1h-nmr测定,根据反应性取代基的马来酰亚胺基或芳与环海藻酸的质子积分值的比,以“mol%(nmr积分比)”记载。
(实施例1)
导入2-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)乙基氨基的海藻酸(al-ex-1)的合成
[化56]
<步骤1>
叔丁基(2-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)乙基)氨基甲酸酯的合成
[化57]
将马来酸酐(600mg)悬浮于乙醇(6.0ml),在冰水冷却下加入叔丁基(2-氨基乙基)氨基甲酸酯(1.03g)和三乙基胺(0.90ml)的乙醇(3.0ml)溶液。将反应液在室温下搅拌2小时后,减压馏去乙醇。将残渣溶解于乙酸酐(6.0ml),加入乙酸钠(502mg),在70℃下搅拌1.5小时。加入乙酸乙酯(25ml)和水(10ml),分液。将有机层依次用饱和小苏打水(10ml、3次)、饱和食盐水(10ml)洗涤后,用无水硫酸钠干燥,减压馏去溶剂。将残渣用硅胶柱色谱(庚烷~50%乙酸乙酯/庚烷)纯化。将所得油状物用庚烷(20ml)磨碎。滤取固体,用庚烷洗涤后,减压干燥,以白色固体形式得到标题化合物(1.01g)。
nmr数据(cdcl3)(δ:ppm):6.71(2h、s)、4.72(1h、brs)、3.66(2h、t、j=6hz)、3.33(2h、q、j=6hz)、1.40(9h、s)。
<步骤2>
1-(2-氨基乙基)-1h-吡咯-2,5-二酮盐酸盐的合成
[化58]
在(实施例1)<步骤1>中得到的化合物(500mg)中加入4当量浓度-氯化氢乙酸乙酯溶液(5.0ml),在室温下搅拌1.5小时。加入乙酸乙酯(5.0ml)后,滤取沉淀,用乙酸乙酯洗涤。将所得吸湿性固体悬浮于乙酸乙酯,减压馏去乙酸乙酯后,减压干燥,以白色固体形式得到标题化合物(328mg)。
nmr数据(d2o)(δ:ppm):6.86(2h、s)、3.80(2h、t、j=6hz)、3.20(2h、t、j=6hz)。
<步骤3>
导入2-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)乙基氨基的海藻酸(al-ex-1)的合成
[化59]
在调整为1重量%的海藻酸钠(株式会公司キミカ制、alg-2)水溶液(20ml)中加入(实施例1)<步骤2>中得到的化合物(36mg)、4-(4、6-二甲氧基-1,3,5-三嗪-2-基)-4-甲基吗啉鎓氯化物(84mg)、1摩尔浓度-小苏打水(252μl),在30℃下搅拌3小时。加入氯化钠(200mg)后,加入乙醇(40ml),在室温下搅拌30分钟。滤取所得沉淀,用乙醇洗涤后,减压干燥,以白色固体形式得到标题化合物(183mg)。
反应性基团的导入率为5.3mol%(nmr积分比)。
分子量在261万da~1万9千da洗脱为宽范围,重均分子量计算为146万da。
(实施例2)
导入2-(2-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)乙氧基)乙基氨基的海藻酸(al-ex-2)的合成
[化60]
<步骤1>
叔丁基(2-(2-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)乙氧基)乙基)氨基甲酸酯的合成
[化61]
将1h-吡咯-2,5-二酮(0.7g)、叔丁基(2-(2-羟基乙氧基)乙基)氨基甲酸酯(1.0g)、三苯基膦(1.4g)溶于四氢呋喃(20ml)。在盐冰水冷却下滴加偶氮二羧酸二异丙基酯(1.9mol/l-甲苯溶液、2.8ml)后,在冰水冷却下搅拌30分钟。在室温下搅拌1小时后,加入乙酸乙酯(20ml)和水(10ml),分液。将有机层用饱和食盐水洗涤后,用无水硫酸钠干燥,减压馏去溶剂。将残渣用硅胶柱色谱(庚烷~乙酸乙酯)纯化后,减压干燥,以淡黄色油状物形式得到标题化合物(0.5g)。
nmr数据(cdcl3)(δ:ppm):6.71(2h、s)、4.87(1h、brs)、3.72(2h、t、j=6hz)、3.59(2h、t、j=6hz)、3.49(2h、t、j=5hz)、3.26(2h、q、j=5hz)、1.44(9h、s)。
<步骤2>
1-(2-(2-氨基乙氧基)乙基)-1h-吡咯-2,5-二酮三氟乙酸盐的合成
[化62]
在冰水冷却下在(实施例2)<步骤1>中得到的化合物(0.5g)中加入三氟乙酸(2.3ml),在室温下搅拌1小时。加入二异丙基醚(11.3ml),在室温下搅拌30分钟后,滤取析出的固体,用二异丙基醚洗涤。将所得吸湿性固体悬浮于二异丙基醚,馏去溶剂后,减压干燥,以淡黄色固体形式得到标题化合物(0.3g)。
nmr数据(dmso-d6)(δ:ppm):7.73(3h、brs)、7.04(2h、s)、3.63-3.53(6h、m)、2.98-2.89(2h、m)。
<步骤3>
导入2-(2-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)乙氧基)乙基氨基的海藻酸(al-ex-2)的合成
[化63]
使用调整为1重量%的海藻酸钠(株式会公司キミカ制、alg-2)水溶液(20ml)和(实施例2)<步骤2>中得到的化合物(60mg),进行与(实施例1)<步骤3>相同的操作,以白色固体形式得到标题化合物(183mg)。
反应性基团的导入率为4.4mol%(nmr积分比)。
分子量在273万da~1万1千da以宽范围洗脱,重均分子量计算为144万da。
(实施例3)
导入2-(2-(2-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)乙氧基)乙氧基)乙基氨基的海藻酸(al-ex-3)的合成
[化64]
<步骤1>
叔丁基(2-(2-(2-氨基乙氧基)乙氧基)乙基)氨基甲酸酯的合成
[化65]
在冰水冷却下花费4.75小时将二碳酸二叔丁基酯(3.0g)的二氯甲烷(37.5ml)溶液滴加在2,2’-(乙烷-1,2-二基双(氧基))乙烷-1-胺)(3.2g)和三乙基胺(11.5ml)的二氯甲烷(30.0ml)溶液中后,在室温下搅拌18.5小时。将反应液减压浓缩,向残渣中加入二氯甲烷(30ml)后,过滤除去不溶物。将滤液依次用水(10ml)、饱和食盐水(10ml)洗涤,用无水硫酸钠干燥后,减压馏去溶剂。将残渣减压干燥,以无色油状物形式得到标题粗化合物(2.7g)。
nmr数据(cdcl3)(δ:ppm):5.15(1h、brs)、3.63-3.60(4h、m)、3.55(2h、t、j=5hz)、3.52(2h、t、j=5hz)、3.32(2h、q、j=5hz)、2.88(2h、t、j=5hz)、1.45(9h、s)。
<步骤2>
叔丁基(2-(2-(2-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)乙氧基)乙氧基)乙基)氨基甲酸酯的合成
[化66]
将(实施例3)<步骤1>中得到的化合物(500mg)、无水马来酸(217mg)悬浮于乙醇(5.0ml),在室温下搅拌30分钟。减压馏去乙醇,将残渣用硅胶柱色谱(庚烷~乙酸乙酯)纯化,得到酰胺体(423mg)。在所得无色油状物和乙酸钠(100mg)中加入乙酸酐(4.2ml),在40℃下搅拌1小时后,在60℃下搅拌1小时、在80℃下搅拌1.5小时、在100℃下搅拌2小时。在反应液中加入乙酸乙酯(25ml)、水(10ml),分液。将有机层依次用饱和小苏打水(10ml)、水(10ml)、饱和食盐水(5ml)洗涤后,用无水硫酸钠干燥,减压浓缩。将残渣用硅胶柱色谱(庚烷~80%乙酸乙酯/庚烷)纯化,以无色油状物形式得到标题化合物(275mg)。
nmr数据(cdcl3)(δ:ppm):6.70(2h、s)、5.01(1h、brs)、3.76-3.72(2h、m)、3.67-3.63(2h、m)、3.61-3.58(2h、m)、3.57-3.54(2h、m)、3.50(2h、t、j=5hz)、3.29(2h、q、j=5hz)、1.45(9h、s)。
<步骤3>
2-(2-(2-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)乙氧基)乙氧基)乙基胺三氟乙酸盐的合成
[化67]
在冰水冷却下在(实施例3)<步骤2>中得到的化合物(275mg)中加入三氟乙酸(1.9ml),在室温下搅拌15分钟。将反应液减压浓缩,将残渣用硅胶柱色谱(乙酸乙酯~30%甲醇/乙酸乙酯)纯化,以无色油状物形式得到标题化合物(231mg)。
nmr数据(cdcl3)(δ:ppm):8.22(3h、brs)、6.74(2h、s)、3.75-3.71(4h、m)、3.64-3.57(6h、m)、3.19(2h、t、j=5hz)。
<步骤4>
导入2-(2-(2-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)乙氧基)乙氧基)乙基氨基的海藻酸(al-ex-3)的合成
[化68]
使用调整为1重量%的海藻酸钠(株式会公司キミカ制、alg-2)水溶液(20ml)和(实施例3)<步骤3>中得到的化合物(69mg),进行与(实施例1)<步骤3>相同的操作,以白色固体形式得到标题化合物(145mg)。
反应性基团的导入率为3.7mol%(nmr积分比)。
分子量在272万da~1万1千da以宽范围洗脱,重均分子量计算为144万da。
(实施例4)
导入s-(4-(2-氨基乙基)氨基甲酰基)苯甲基)乙酰基硫醇基的海藻酸(al-ex-4)的合成
[化69]
<步骤1>
叔丁基(2-(4-(氯甲基)苯甲酰胺基)乙基)氨基甲酸酯的合成
[化70]
将4-(氯甲基)苯甲酰氯(2.0g)溶于四氢呋喃(10.0ml),在冰水冷却下滴加叔丁基(2-氨基乙基)氨基甲酸酯(1.7g)和二异丙基乙基胺(3.7ml)的四氢呋喃(10.0ml)溶液,在室温下搅拌1.5小时。在反应液中加入乙酸乙酯(30ml)和水(10ml),分液。将有机层依次用半饱和小苏打水(10ml)、水(10ml)、饱和食盐水(5ml)洗涤,用无水硫酸钠干燥后,减压浓缩。将残渣用叔丁基甲基醚磨碎后,滤取所得固体,用叔丁基甲基醚洗涤,以白色固体形式得到标题化合物(2.9g)。
nmr数据(cdcl3)(δ:ppm):7.81(2h、d、j=8hz)、7.44(2h、d、j=8hz)、7.24(1h、brs)、4.96(1h、brs)、4.60(2h、s)、3.56(2h、q、j=5hz)、3.45-3.38(2h、m)、1.43(9h、s)。
<步骤2>
s-(4-(2-(叔丁氧基羰基)氨基)乙基)氨基甲酰基)苯甲基)乙酰基硫醇的合成
[化71]
将(实施例4)<步骤1>中得到的化合物(1.20g)悬浮于乙腈(24.0ml)。加入硫代乙酸钾(0.53g),在室温下搅拌30分钟。在反应液中加入乙酸乙酯(50ml)、水(20ml),分液。将有机层依次用水(20ml)、饱和食盐水(10ml)洗涤,用无水硫酸钠干燥后,减压浓缩。将残渣用叔丁基甲基醚磨碎后,滤取固体,用叔丁基甲基醚洗涤。将所得固体在40℃下减压干燥,以白色固体形式得到标题化合物(1.27g)。
nmr数据(cdcl3)(δ:ppm):7.74(2h、d、j=8hz)、7.33(2h、d、j=8hz)、7.15(1h、brs)、4.96(1h、brs)、4.13(2h、s)、3.54(2h、q、j=5hz)、3.43-3.36(2h、m)、2.36(3h、s)、1.43(9h、s)。
<步骤3>
s-(4-(2-氨基乙基)氨基甲酰基)苯甲基)乙酰基硫醇盐酸盐的合成
[化72]
在冰水冷却下在(实施例4)<步骤2>中得到的化合物(0.60g)中加入4当量浓度-氯化氢/1,4-二氧杂环己烷(4.2ml),在室温下搅拌30分钟。追加4当量浓度-氯化氢/1,4-二氧杂环己烷(2.1ml),在室温下进一步搅拌30分钟。在反应液中加入二异丙基醚(12.6ml),滤取所得沉淀,用二异丙基醚洗涤后,减压干燥,以白色固体形式得到标题化合物(0.46g)。
nmr数据(dmso-d6)(δ:ppm):8.65(1h、t、j=6hz)、7.85(3h、brs)、7.82(2h、d、j=8hz)、7.38(2h、d、j=8hz)、4.16(2h、s)、3.49(2h、q、j=6hz)、2.97(2h、t、j=6hz)、2.36(3h、s)。
<步骤4>
导入s-(4-(2-氨基乙基)氨基甲酰基)苯甲基)乙酰基硫醇基的海藻酸(al-ex-4)的合成
[化73]
使用调整为1重量%的海藻酸钠(株式会公司キミカ制、alg-2)水溶液(20ml)和(实施例4)<步骤3>中得到的化合物(58mg),进行与(实施例1)<步骤3>相同的操作,以白色固体形式得到标题化合物(189mg)。
反应性基团的导入率为5.6mol%(nmr积分比)。
分子量在277万da~1万4千da以宽范围洗脱,重均分子量计算为142万da。
(实施例5)
导入s-(4-(3-((3-氨基丙基)氨基)-3-氧代丙基)苯甲基)乙酰基硫醇基的海藻酸(al-ex-5)的合成
[化74]
<步骤1>
4-(3-((3-((叔丁氧基羰基)氨基)丙基)氨基)-3-氧代丙基)苯甲酸甲基酯的合成
[化75]
将3-(4-(甲氧基羰基)苯基)丙烷酸(1.15g)、叔丁基(3-氨基丙基)氨基甲酸酯(0.96g)溶于甲醇(11.5ml)。加入4-(4,6-二甲氧基-1,3,5-三嗪-2-基)-4-甲基吗啉鎓氯化物(2.14g),在室温下搅拌2小时、在40℃下搅拌1小时。在反应液中加入乙酸乙酯(20ml)、水(20ml),分液,将水层用乙酸乙酯(10ml)提取。合并有机层,依次用半饱和小苏打水(10ml)、水(10ml)、饱和食盐水(5ml)洗涤后,用无水硫酸钠干燥,减压馏去溶剂。将残渣用硅胶柱色谱(10%乙酸乙酯/庚烷~乙酸乙酯)纯化,以无色油状物形式得到标题化合物(0.76g)。
nmr数据(cdcl3)(δ:ppm):7.95(2h、d、j=8hz)、7.28(2h、d、j=8hz)、6.19(1h、brs)、4.79(1h、brs)、3.90(3h、s)、3.25(2h、q、j=6hz)、3.08-3.01(4h、m)、2.51(2h、t、j=8hz)、1.57-1.49(2h、m)、1.43(9h、s)。
<步骤2>
叔丁基(3-(3-(4-(羟基甲基)苯基)丙烷酰胺)丙基)氨基甲酸酯的合成
[化76]
将(实施例5)<步骤1>中得到的化合物(560mg)溶于四氢呋喃(11.2ml)。花费5分钟加入氢化铝锂(146mg),在室温下搅拌1小时。在冰水冷却下,加入饱和硫酸钠水溶液(50滴)后,在相同温度下搅拌1小时。过滤除去析出的不溶物,用四氢呋喃洗涤。将滤液减压浓缩,以无色油状物形式得到标题化合物(569mg)。
nmr数据(cdcl3)(δ:ppm):7.28(2h、d、j=8hz)、7.20(2h、d、j=8hz)、5.95(1h、brs)、4.79(1h、brs)、4.65(2h、s)、3.23(2h、q、j=6hz)、2.99-2.92(4h、m)、2.48(2h、t、j=7hz)、1.54-1.47(2h、m)、1.44(9h、s)。
<步骤3>
叔丁基(3-(3-(4-(氯甲基)苯基)丙烷酰胺)丙基)氨基甲酸酯的合成
[化77]
将(实施例5)<步骤2>中得到的化合物(400mg)溶于四氢呋喃(8.0ml)。加入对甲苯磺酰氯(272mg)、n、n-二甲基-4-氨基吡啶(15mg)、三乙基胺(0.33ml),在70℃下搅拌6小时。在反应液中加入乙酸乙酯(25ml)、水(10ml),分液,将水层用乙酸乙酯(5ml)提取。合并有机层,依次用半饱和小苏打水(10ml)、水(10ml)、饱和食盐水(5ml)洗涤后,用无水硫酸钠干燥,减压浓缩。将残渣用叔丁基甲基醚/庚烷磨碎,滤取所得固体后,用庚烷洗涤,以浅米色固体形式得到标题化合物(224mg)。
nmr数据(cdcl3)(δ:ppm):7.30(2h、d、j=8hz)、7.20(2h、d、j=8hz)、6.13(1h、brs)、4.81(1h、brs)、4.56(2h、s)、3.28-3.21(2h、m)、3.04(2h、q、j=6hz)、2.97(2h、t、j=8hz)、2.48(2h、t、j=8hz)、1.56-1.47(2h、m)、1.43(9h、s)。
<步骤4>
s-(4-(3-((3-((叔丁氧基羰基)氨基)丙基)氨基)-3-氧代丙基)苯甲基)乙酰基硫醇的合成
[化78]
将(实施例5)<步骤3>中得到的化合物(224mg)悬浮于乙腈(4.5ml)。加入硫代乙酸钾(87mg),在室温下搅拌30分钟。在反应液中加入乙酸乙酯(20ml)、水(10ml),分液。将有机层依次用水(10ml)、饱和食盐水(5ml)洗涤,用无水硫酸钠干燥后,减压浓缩。将残渣用硅胶柱色谱(10%乙酸乙酯/庚烷~乙酸乙酯)纯化,以白色固体形式得到标题化合物(189mg)。
nmr数据(cdcl3)(δ:ppm):7.19(2h、d、j=8hz)、7.13(2h、d、j=8hz)、6.07(1h、brs)、4.82(1h、brs)、4.08(2h、s)、3.25(2h、q、j=6hz)、3.04(2h、q、j=6hz)、2.94(2h、t、j=8hz)、2.46(2h、t、j=8hz)、2.34(3h、s)、1.56-1.49(2h、m)、1.43(9h、s)。
<步骤5>
s-(4-(3-((3-氨基丙基)氨基)-3-氧代丙基)苯甲基)乙酰基硫醇盐酸盐的合成
[化79]
使用(实施例5)<步骤4>中得到的化合物(189mg),进行与(实施例4)<步骤3>相同的操作,以白色固体形式得到标题化合物(140mg)。
nmr数据(dmso-d6)(δ:ppm):8.03(1h、t、j=6hz)、7.79(3h、brs)、7.18(2h、d、j=8hz)、7.12(2h、d、j=8hz)、4.07(2h、s)、3.09(2h、q、j=6hz)、2.77(2h、t、j=8hz)、2.75-2.66(2h、m)、2.38-2.33(2h、m)、2.34(3h、s)、1.68-1.60(2h、m)。
<步骤6>
导入s-(4-(3-((3-氨基丙基)氨基)-3-氧代丙基)苯甲基)乙酰基硫醇基的海藻酸(al-ex-5)的合成
[化80]
使用调整为1重量%的海藻酸钠(株式会公司キミカ制、alg-2)水溶液(20ml)和(实施例5)<步骤5>中得到的化合物(67mg),进行与(实施例1)<步骤3>相同的操作,以白色固体形式得到标题化合物(189mg)。
反应性基团的导入率为4.2mol%(nmr积分比)。
分子量在261万da~2万1千da以宽范围洗脱,重均分子量计算为142万da。
(实施例7-1)
导入2-(n-(4-(巯基甲基)苯甲酰胺基))乙基氨基的海藻酸(al-ex-7-1)水溶液的制备
[化81]
将(实施例4)<步骤4>中得到的化合物(160mg)溶于水(8.0ml),加入1当量浓度-氢氧化钠水溶液(112μl),在25℃下搅拌2小时,制备标题化合物的2重量%溶液。进行乙醇的沉淀处理时,形成凝胶状,因此直接将溶液用于试验。部分进行乙醇处理,用nmr确认乙酰基的消失。
(实施例8)
导入2-氨基-n-(2-(2、5-二氧代-2、5-二氢-1h-吡咯-1-基)乙基)乙酰胺基的海藻酸(al-ex-8)的合成
[化82]
<步骤1>
叔丁基(2-((2-(2、5-二氧代-2、5-二氢-1h-吡咯-1-基)乙基)氨基)-2-氧代乙基)氨基甲酸酯的合成
[化83]
相对市售的1-(2-氨基乙基)-1h-吡咯-2、5-二酮盐酸盐[casno.134272-64-3](92.43mg)和水(750μl)的混合物在室温下加入1摩尔浓度-小苏打水(578.5μl)。相对该混合物在室温下加入市售的2、5-二氧代吡咯烷-1-基(叔丁氧基羰基)甘氨酸酯[cas:3392-07-2](150mg)的四氢呋喃(1500μl)溶液,在相同温度下搅拌30分钟。反应结束后,加入乙酸乙酯(10ml)和水(5ml),分离。将有机层用无水硫酸钠干燥,过滤后,在减压下浓缩。将粗产物用硅胶柱色谱(25%乙酸乙酯/庚烷~100%乙酸乙酯、乙酸乙酯~60%甲醇/乙酸乙酯)纯化,以无色油状化合物形式得到标题化合物(74mg)。
nmr数据(cdcl3)(δ:ppm):6.72(2h、s)、6.59(1h、brs)、5.18(1h、brs)、3.74(2h、d、j=6hz)、3.72-3.68(2h、m)、3.50-3.46(2h、m)、1.45(9h、s)。
<步骤2>
2-氨基-n-(2-(2、5-二氧代-2、5-二氢-1h-吡咯-1-基)乙基)乙酰胺三氟乙酸盐的合成
[化84]
相对(实施例8)<步骤1>中得到的化合物(0.074g)和二氯甲烷(0.22ml)的混合物在冰冷却搅拌下加入三氟乙酸(0.52ml),在室温下搅拌2小时。反应结束后,浓缩反应液,加入二异丙基醚(20ml)。由于形成胶状化合物,因此将混合物在减压下浓缩,干燥,由此以淡黄色胶状化合物形式得到标题粗化合物(0.097g)。
nmr数据(dmso-d6)(δ:ppm):8.45(1h、t、j=6hz)、8.00(3h、brs)、7.03(2h、s)、3.48(2h、t、j=6hz)、3.43(2h、q、j=6hz)、3.29(2h、q、j=6hz)。
<步骤3>
导入2-氨基-n-(2-(2、5-二氧代-2、5-二氢-1h-吡咯-1-基)乙基)乙酰胺基的海藻酸(al-ex-8)的合成
[化85]
在室温下在调整为1重量%的海藻酸钠(株式会公司キミカ制、alg-2)水溶液(29.7ml)中加入4-(4、6-二甲氧基-1、3、5-三嗪-2-基)-4-甲基吗啉鎓氯化物(68.6mg)、1摩尔浓度-小苏打水(68.6μl)。接着,在相同温度下缓慢加入(实施例8)<步骤2>中得到的化合物(21.4mg)、水(1ml)和乙醇(1ml)的混合物,在40℃下搅拌4小时。加入氯化钠(300mg)后,加入乙醇(59.3ml),在室温下搅拌30分钟。滤取所得沉淀,用乙醇洗涤后,减压干燥,以白色絮状化合物形式得到标题化合物(221.3mg)。
交联基团的导入率为4.8mol%(nmr积分比)。
分子量在273万da~2千da以宽范围洗脱,重均分子量计算为136万da。
(实施例9)
导入(s)-2-氨基-n-(2-(2、5-二氧代-2、5-二氢-1h-吡咯-1-基)乙基)-3-苯基丙烷酰胺基的海藻酸(al-ex-9)的合成
[化86]
<步骤1>
叔丁基(s)-(1-((2-(2、5-二氧代-2、5-二氢-1h-吡咯-1-基)乙基)氨基)-1-氧代-3-苯基丙烷-2-基)氨基甲酸酯的合成
[化87]
相对市售的1-(2-氨基乙基)-1h-吡咯-2、5-二酮盐酸盐[cas:134272-64-3](100mg)、市售的(叔丁氧基羰基)-l-苯丙氨酸[casno.13734-34-4](150.23mg)和二氯甲烷(1ml)的混合物,在冰冷却搅拌下加入三乙基胺(78.9μl)。对该混合物,在相同温度下加入n、n’-二环己基碳二酰亚胺(116.8mg),在室温下搅拌30分钟。反应结束后,用乙酸乙酯(20ml)稀释,过滤悬浮液。将粗产物用硅胶柱色谱(12%乙酸乙酯/庚烷~100%乙酸乙酯)纯化,以白色无定形形式得到标题化合物(108mg)。
nmr数据(cdcl3)(δ:ppm):7.30-7.27(2h、m)、7.23-7.17(3h、m)、6.68(2h、s)、6.19(1h、brs)、4.91(1h、brs)、4.30-4.26(1h、m)、3.66-3.53(2h、m)、3.50-3.41(1h、m)、3.36-3.30(1h、m)、3.08-2.98(2h、m)、1.39(9h、s)。
<步骤2>
(s)-2-氨基-n-(2-(2、5-二氧代-2、5-二氢-1h-吡咯-1-基)乙基)-3-苯基丙烷酰胺三氟乙酸盐的合成
[化88]
相对(实施例9)<步骤1>中得到的化合物(0.1g)和二氯甲烷(1.3ml)的混合物,在冰冷却搅拌下,加入三氟乙酸(0.7ml),在室温下搅拌30分钟。反应结束后,将反应液在减压下浓缩,加入二异丙基醚(20ml)。过滤悬浮液,以白色固体形式得到标题化合物(0.12g)。
nmr数据(dmso-d6)(δ:ppm):8.57(1h、t、j=6hz)、8.07(3h、brs)、7.36-7.33(2h、m)、7.30-7.22(3h、m)、7.05(2h、s)、3.85(1h、dd、j=8、6hz)、3.45-3.40(3h、m)、3.18-3.13(1h、m)、3.01(1h、dd、j=14、5hz)、2.83(1h、dd、j=14、9hz)。
<步骤3>
导入(s)-2-氨基-n-(2-(2、5-二氧代-2、5-二氢-1h-吡咯-1-基)乙基)-3-苯基丙烷酰胺基的海藻酸(al-ex-9)的合成
[化89]
使用调整为1重量%的海藻酸钠(株式会公司キミカ制、alg-2)水溶液(29.7ml)和(实施例9)<步骤2>中得到的化合物(27.5mg),进行与(实施例8)<步骤3>相同的操作,以白色絮状化合物形式得到标题化合物(264.8mg)。
交联基团的导入率为6.0mol%(nmr积分比)。
分子量在277万da~6千da以宽范围洗脱,重均分子量计算为146万da。
(实施例10)
导入(s)-2-(2-氨基乙酰胺)-n-(2-(2、5-二氧代-2、5-二氢-1h-吡咯-1-基)乙基)-3-苯基丙烷酰胺基的海藻酸(al-ex-10)的合成
[化90]
<步骤1>
(叔丁氧基羰基)甘氨酰-l-苯丙氨酸的合成
[化91]
相对市售的l-苯丙氨酸[cas:63-91-2](0.12g)和水(1ml)的混合物,在室温下加入1摩尔浓度-小苏打水(0.73ml)。相对该混合物在室温下加入市售的2、5-二氧代吡咯烷-1-基(叔丁氧基羰基)甘氨酸酯[cas:3392-07-2](0.2g)的四氢呋喃(4ml)溶液,在相同温度下搅拌。1小时30分钟后,进一步加入2、5-二氧代吡咯烷-1-基(叔丁氧基羰基)甘氨酸酯(0.02g),在室温下搅拌30分钟。反应结束后加入乙酸乙酯(10ml)和1当量浓度-盐酸(3ml),分离。有机层依次用水(5ml)和饱和食盐水(5ml)洗涤,用无水硫酸钠干燥,过滤后,在减压下浓缩。将粗产物用硅胶柱色谱(25%乙酸乙酯/庚烷~100%乙酸乙酯、乙酸乙酯~20%甲醇/乙酸乙酯)纯化,以白色无定形形式得到标题化合物(0.21g)。
nmr数据(cdcl3)(δ:ppm):7.31-7.20(3h、brm)、7.16-7.13(2h、m)、6.65(1h、brs)、5.26(1h、brs)、4.86(1h、brs)、3.88(1h、dd、j=17、7hz)、3.68(1h、dd、j=17、6hz)、3.24-3.05(2h、m)、1.44(9h、s)。
<步骤2>
叔丁基(s)-(2-((1-((2-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)乙基)氨基)-1-氧代-3-苯基丙烷-2-基)氨基)-2-氧代乙基)氨基甲酸酯的合成
[化92]
相对市售的1-(2-氨基乙基)-1h-吡咯-2,5-二酮盐酸盐[cas:134272-64-3](114mg)、(实施例10)<步骤1>中得到的化合物(208mg)和二氯甲烷(2080μl)的混合物,在冰冷却搅拌下加入三乙基胺(90μl)。相对该混合物,在相同温度下加入n,n’-二环己基碳二酰亚胺(133.1mg),在室温下搅拌1小时30分钟。反应结束后,用乙酸乙酯(20ml)稀释,过滤悬浮液。将粗产物用硅胶柱色谱(25%乙酸乙酯/庚烷~100%乙酸乙酯、乙酸乙酯~20%甲醇/乙酸乙酯)纯化。将回収的组分在减压下浓缩,在叔丁基甲基醚(20ml)中溶解。将该溶液依次用饱和小苏打水(5ml)、水(5ml)洗涤2次、用饱和食盐水(5ml)洗涤,用无水硫酸钠干燥。将有机层在减压下浓缩,以白色无定形形式得到标题化合物(220mg)。
nmr数据(cdcl3)(δ:ppm):7.29-7.27(2h、m)、7.24-7.16(3h、m)、6.67(2h、s)、6.47(1h、brs)、6.40(1h、d、j=8hz)、5.14(1h、brs)、4.66-4.60(1h、m)、3.85-3.45(5h、m)、3.29-3.23(1h、m)、3.09(2h、d、j=7hz)、1.44(9h、s)。
<步骤3>
(s)-2-(2-氨基乙酰胺)-n-(2-(2、5-二氧代-2、5-二氢-1h-吡咯-1-基)乙基)3-苯基丙烷酰胺三氟乙酸盐的合成
[化93]
使用(实施例10)<步骤2>中得到的化合物(0.22g),进行与(实施例9)<步骤2>相同的操作,以白色固体形式得到标题化合物(0.25g)。
nmr数据(dmso-d6)(δ:ppm):8.67(1h、d、j=8hz)、8.33(1h、t、j=6hz)、7.89(3h、brs)、7.29-7.24(2h、m)、7.23-7.17(3h、m)、7.03(2h、s)、4.46-4.40(1h、m)、3.48-3.41(3h、m)、3.38-3.28(2h、m)、3.17-3.11(1h、m)、2.94(1h、dd、j=14、4hz)、2.67(1h、dd、j=14、10hz)。
<步骤4>
导入(s)-2-(2-氨基乙酰胺)-n-(2-(2、5-二氧代-2、5-二氢-1h-吡咯-1-基)乙基)-3-苯基丙烷酰胺基的海藻酸(al-ex-10)的合成
[化94]
使用调整为1重量%的海藻酸钠(株式会公司キミカ制、alg-2)水溶液(49.4ml)和(实施例10)<步骤3>中得到的化合物(52.4mg),进行与(实施例8)<步骤3>相同的操作,以白色絮状化合物形式得到标题化合物(485mg)。
交联基团的导入率为5.3mol%(nmr积分比)。
分子量在287万da~2万da以宽范围洗脱,重均分子量计算为143万da。
<反应性基团的导入率测定>
反应性基团的导入率是指将作为海藻酸的重复单元的每糖醛酸单糖单元所导入的反应性基团的数以百分数表示的值。算出导入率所必需的海藻酸的量通过利用校正曲线得到的咔唑硫酸法进行测定,反应性基团的量通过利用校正曲线的吸光度测定法进行测定。
<分子量的测定>
称量实施例中得到的导入交联基团的海藻酸固体,添加至包含0.15mol/l的nacl的10mmol/l磷酸缓冲液(ph7.4),在室温下搅拌1小时以上而溶解,制备0.2%溶液。将该溶液通过孔径0.45μm的聚醚砜制minisarthighflow过滤器(sartorius公司)而除去不溶物后,将该200μl供于superose6increase10/300gl柱(ge医疗科学公司),实施凝胶过滤。凝胶过滤使用aktaexplorer10s作为色谱装置,使用包含0.15mol/alacl的10mmol/l磷酸缓冲液(ph7.4)作为展开溶剂,在室温下流速为0.8ml/mim的条件下实施。检测220nm或240nm的吸収并制作各样品的色谱图。此外作为另一方法,检测215nm的吸収。所得色谱图的峰解析使用unicorn5.31软件(ge医疗科学公司)进行。
导入交联基团的海藻酸的分子量使用蓝色葡聚糖(分子量200万da、sigma公司)、甲状腺球蛋白(分子量66.9万da、ge医疗科学公司)、铁蛋白(分子量44万da、ge医疗科学公司)、醛缩酶(分子量15.8万da、ge医疗科学公司)、伴白蛋白(分子量7.5万da、ge医疗科学公司)、卵白蛋白(分子量4.4万da、ge医疗科学公司)、核糖核酸酶a(分子量1.37万da、ge医疗科学公司)和抑肽酶(分子量6500da、ge医疗科学公司)作为标准品,由在相同条件下进行凝胶过滤时的各成分的280nm下的吸収峰的液体量和分子量制作校正曲线。从蓝色葡聚糖至铁蛋白、从铁蛋白至抑肽酶制作2种校正曲线。使用该校正曲线,先计算得到的色谱的洗脱时间i中的分子量(mi)。接着,读取洗脱时间i中的吸光度并作为hi,根据这些数据由下述求出重均分子量(mw)。
[数学式1]
导入交联基团前的海藻酸的分子量如以下确定。即,考虑干燥减重,称量各海藻酸,加入超纯水,制成1%水溶液。接着,以海藻酸浓度达到0.2%、溶液组成为包含0.15mol/l的nacl的10mmol/l磷酸缓冲液(ph7.4)的方式进行稀释。将不溶物利用孔径0.22μm的亲水性pvdf制mylexgv33过滤器(merck-millipore公司)除去后,将200μl供于凝胶过滤,在与导入交联基团的海藻酸相同的条件下实施凝胶过滤。检测利用差示折射计实施。此外,作为另一方法,通过孔径0.45μm的聚醚砜制minisarthighflow过滤器(sartorius公司)除去不溶物。
导入交联基团前的海藻酸的重均分子量利用与导入交联基团的海藻酸的分子量的计算方法相同的方法确定。其中,hi根据差示屈折计的数据计算。
实施例1~5、和实施例7-1中使用的导入交联基团前的海藻酸(alg-2)的分子量在260万da~14.5万da以宽范围洗脱,重均分子量计算为146万da。
实施例8~10中使用的导入交联基团前的海藻酸(alg-2)的分子量在9600da~251万da以宽范围洗脱,重均分子量计算为138万da。
<凝胶稳定性的测定>
将(实施例2)<步骤3>中得到的海藻酸衍生物(al-ex-2)、或(实施例3)<步骤4>中得到的海藻酸衍生物(al-ex-3)分别以浓度达到1%的方式溶于水而得到海藻酸水溶液(2)和海藻酸水溶液(3)。进一步在(实施例7-1)中得到的2重量%的海藻酸衍生物(al-ex-7-1)溶液中等量添加磷酸缓冲生理食盐水(pbs)而形成1重量%,得到海藻酸水溶液(7-1)。
等量混合海藻酸水溶液(2)和海藻酸水溶液(7-1),将该混合得到的水溶液加入装有18规格的注射针的注射筒,将该注射筒设置在设定为流速1ml/分钟的注射泵上,滴加在浓度为300mmol/l的氯化钙溶液中30秒钟,搅拌5分钟,得到海藻酸凝胶。将该凝胶用10ml的pbs洗涤1次后,在pbs中在37℃下静置10分钟,进行化学交联,得到化学交联海藻酸凝胶。在该凝胶中添加20ml的pbs,在37℃下振荡,经时地回收水溶液,补充与回収的量相同量的pbs。试验结束后,在试验溶液中添加2μl海藻酸裂解酶(ニッポンジーン、319-08261),在37℃振荡1小时使凝胶完全崩解,回收水溶液。通过咔唑硫酸法测定回収的水溶液中的海藻酸浓度,将用已经回收的海藻酸浓度校正各时刻的水溶液中海藻酸浓度而得的值除以由全部时刻的海藻酸浓度和试验结束后的海藻酸浓度算出的总海藻酸浓度而得的值以百分数表示,将得到的值作为崩解率,作为凝胶稳定性的指标。此外,对于使用海藻酸水溶液(3)和海藻酸水溶液(7-1)按照上述的方法得到的海藻酸凝胶,也测定凝胶稳定性。
将结果示于图1。
作为对照使用的由海藻酸(alg-2)制成的海藻酸凝胶在8小时基本溶解,与此相对,本实施例的将导入交联基团的海藻酸衍生物交联而得到的交联海藻酸结构体(将海藻酸衍生物(al-ex-2)/海藻酸衍生物(al-ex-7-1)交联而得到的交联海藻酸结构体、和将海藻酸衍生物(al-ex-3)/海藻酸衍生物(al-ex-7-1)交联而得到的交联海藻酸结构体)的稳定性均提高。
<凝胶稳定性的测定(2)>
将实施例8中得到的海藻酸衍生物(al-ex-8)、实施例9中得到的海藻酸衍生物(al-ex-9)、和实施例10中得到的海藻酸衍生物(al-ex-10)以浓度达到0.5%的方式溶于水,得到海藻酸水溶液(8)、海藻酸水溶液(9)、和海藻酸水溶液(10)。进一步在利用与实施例7-1相同的方法得到的2%的导入交联基团的海藻酸衍生物(al-ex-7-1-2)(导入率(nmr积分比)=5.1mol%)中加入磷酸缓冲生理食盐水(pbs)3容量而形成0.5%,得到海藻酸水溶液(7-1-2)。
将海藻酸水溶液(8)、海藻酸水溶液(9)、和海藻酸水溶液(10)的各海藻酸水溶液与海藻酸水溶液(7-1-2)分别以250μl等量混合,添加40ml的浓度为30mmol/l的氯化钙溶液,搅拌5分钟,得到海藻酸凝胶。将该凝胶用10ml的pbs洗涤1次,得到化学交联海藻酸凝胶。在该凝胶中添加19.5ml的pbs,在37℃下振荡,经时地回收水溶液,补充与回収的量相同量的pbs。试验结束后,在试验溶液中添加10μl的海藻酸裂解酶(ニッポンジーン、319-08261),在37℃下整夜振荡使凝胶完全崩解,回收水溶液。通过咔唑硫酸法测定回収的水溶液中的海藻酸浓度,将用已经回收的海藻酸浓度校正各时刻的水溶液中海藻酸浓度而得的值除以由全部时刻的海藻酸浓度和试验结束后的海藻酸浓度算出的总海藻酸浓度而得的值以百分数表示,将得到的值作为崩解率,作为凝胶稳定性的指标。
将结果示于图3。
将海藻酸衍生物(al-ex-9)/海藻酸衍生物(al-ex-7-1-2)交联而得到的交联海藻酸结构体的96小时后的崩解率约为39%,将海藻酸衍生物(al-ex-8)/海藻酸衍生物(al-ex-7-1-2)交联而得到的交联海藻酸结构体的96小时后的崩解率约为40%,并且将海藻酸衍生物(al-ex-10)/海藻酸衍生物(al-ex-7-1-2)交联而得到的交联海藻酸结构体的96小时后的崩解率约为55%,凝胶稳定性的测定(1)的由海藻酸(alg-2)制作的海藻酸凝胶相比,暗示稳定性提高。
<凝胶稳定性的测定(3)>
将利用与(实施例2)<步骤3>相同的方法制造的导入率(nmr积分比)=3.4mol%的导入交联基团的海藻酸衍生物(al-ex-2-1)、实施例8中得到的海藻酸衍生物(al-ex-8)、实施例9中得到的海藻酸衍生物(al-ex-9)、和实施例10中得到的海藻酸衍生物(al-ex-10)以浓度达到0.5%的方式溶于水而得到海藻酸水溶液(2‐1)、海藻酸水溶液(8)、海藻酸水溶液(9)、和海藻酸水溶液(10)。进一步在利用与实施例7-1相同的方法得到的2%的导入交联基团的海藻酸衍生物(al-ex-7-1-2)(导入率(nmr积分比)=5.1mol%)中加入3容量磷酸缓冲生理食盐水(pbs),形成0.5%,得到海藻酸水溶液(7-1-2)。
将海藻酸水溶液(2‐1)、海藻酸水溶液(8)、海藻酸水溶液(9)、和海藻酸水溶液(10)的各海藻酸水溶液与海藻酸水溶液(7-1-2)分别以250μl等量混合,添加40ml浓度为30mmol/l的氯化钙溶液,搅拌5分钟,得到海藻酸凝胶。在该凝胶中添加19.5ml的5mm乙二胺四乙酸二钾盐二水合物(edta・2k)/生理食盐水溶液,在37℃振荡,在24小时后回收水溶液,补充与回収的量相同量的5mmedta・2k/生理食盐水溶液。试验结束后,在试验溶液中添加10μl海藻酸裂解酶(ニッポンジーン、319-08261),在37℃下整夜振荡使凝胶完全崩解,回收水溶液。通过咔唑硫酸法测定回収的水溶液中的海藻酸浓度,将用已经回收的海藻酸浓度校正各时刻的水溶液中海藻酸浓度而得的值除以由全部时刻的海藻酸浓度和试验结束后的海藻酸浓度算出的总海藻酸浓度而得的值以百分数表示,将得到的值作为崩解率,作为凝胶稳定性的指标。
将结果示于图4。
将海藻酸衍生物(al-ex-2-1)/海藻酸衍生物(al-ex-7-1-2)交联而得到的交联海藻酸结构体在24小时后崩解率约为49%,将海藻酸衍生物(al-ex-9)/海藻酸衍生物(al-ex-7-1-2)交联而得到的交联海藻酸结构体在24小时后崩解率约为28%,将海藻酸衍生物(al-ex-8)/海藻酸衍生物(al-ex-7-1-2)交联而得到的交联海藻酸结构体在24小时后崩解率约为32%、并且将海藻酸衍生物(al-ex-10)/海藻酸衍生物(al-ex-7-1-2)交联而得到的交联海藻酸结构体在24小时后崩解率约为32%,除钙交联的海藻酸结构体均可确认具有稳定性。
<凝胶泄漏率的测定(1)>
将(实施例2)<步骤3>中得到的海藻酸衍生物(al-ex-2)、或(实施例3)<步骤4>中得到的海藻酸衍生物(al-ex-3)、或者作为原料海藻酸的未导入反应性基团的海藻酸(alg-2;对照)以浓度达到1%的方式溶于水,添加1/100容量的1当量浓度-碳酸氢钠水溶液,得到海藻酸水溶液(2)、海藻酸水溶液(3)和海藻酸水溶液(alg-2-aq)。进一步在(实施例7-1)中得到的2重量%的海藻酸衍生物(al-ex-7-1)溶液中等量添加用磷酸缓冲生理食盐水(pbs)制备为1mg/ml的分子量200万的异硫氰酸荧光素-葡聚糖(シグマアルドリッチ、fd2000s)而形成1重量%,得到海藻酸水溶液(7-1)。
等量混合海藻酸水溶液(2)和海藻酸水溶液(7-1)、或海藻酸水溶液(3)和海藻酸水溶液(7-1),将该混合得到的水溶液加入装有18规格的注射针的注射筒,将该注射筒设置在设定为流速1ml/分的注射泵上,滴加在浓度为30mmol/l的氯化钙溶液中30秒钟,搅拌约20分钟,得到海藻酸凝胶。将该凝胶用10ml的pbs洗涤1次,得到内包异硫氰酸荧光素-葡聚糖的化学交联海藻酸凝胶。在该凝胶中添加20ml的pbs,在37℃下振荡,经时地回收水溶液。试验结束后,在试验溶液中添加5μl海藻酸裂解酶(ニッポンジーン、319-08261),在37℃下振荡2小时而使凝胶完全崩解,回收水溶液。通过荧光定量法(激发光:485nm、荧光:535nm)测定回収的水溶液中的葡聚糖浓度,将各时刻的葡聚糖浓度除以试验结束后的葡聚糖浓度而得的值以百分数表示,将所得值作为透过率,作为凝胶稳定性的指标。
将结果示于图2。
相对于作为对照使用的由海藻酸(alg-2)制备的凝胶在24小时显示不到40%、在48小时显示约70%的泄漏率,本实施例的将导入交联基团的海藻酸衍生物交联而得到的交联海藻酸结构体(将海藻酸衍生物(al-ex-2)/海藻酸衍生物(al-ex-7-1)交联而得到的交联海藻酸结构体、和将海藻酸衍生物(al-ex-3)/海藻酸衍生物(al-ex-7-1)交联而得到的交联海藻酸结构体)的24小时的泄漏率约为10%、48小时的泄漏率为10~15%左右,稳定性均提高。
<凝胶透过率的测定(2)>
将利用与(实施例2)<步骤3>相同的方法制造的导入率(nmr积分比)=3.4mol%的导入交联基团的海藻酸衍生物(al-ex-2-1)、实施例8中得到的海藻酸衍生物(al-ex-8)、实施例9中得到的海藻酸衍生物(al-ex-9)、和实施例10中得到的海藻酸衍生物(al-ex-10)以浓度达到2.0%的方式溶于水,制备海藻酸水溶液,在该海藻酸水溶液中加入4/5容量的制备为1mg/ml的分子量15万的异硫氰酸荧光素-葡聚糖(シグマアルドリッチ、fd150s)、和2.2容量的水,得到含有0.2mg/ml异硫氰酸荧光素-葡聚糖的0.5%海藻酸水溶液(2‐1)、海藻酸水溶液(8)、海藻酸水溶液(9)、和海藻酸水溶液(10)。进一步在利用与实施例7-1相同的方法得到的2%的导入交联基团的海藻酸衍生物(al-ex-7-1-2)(导入率(nmr积分比)=5.1mol%)中加入3容量磷酸缓冲生理食盐水(pbs),形成0.5%,得到海藻酸水溶液(7-1-2)。
将海藻酸水溶液(2‐1)、海藻酸水溶液(8)、海藻酸水溶液(9)、和海藻酸水溶液(10)的各海藻酸水溶液与海藻酸水溶液(7-1-1-2)分别以250μl等量混合,添加40ml浓度为30mmol/l的氯化钙溶液,搅拌5分钟,得到海藻酸凝胶。将该凝胶用10ml的生理食盐水洗涤1次,得到内包异硫氰酸荧光素-葡聚糖的化学交联海藻酸凝胶。在该凝胶中添加19.5ml的生理食盐水,在37℃下振荡,经时地回收水溶液,补充与回収的量相同量的pbs。试验结束后,在试验溶液中添加10μl海藻酸裂解酶(ニッポンジーン、319-08261),在37℃下振荡3小时以上使凝胶完全崩解,回收水溶液。通过荧光定量法(激发光:485nm、荧光:535nm)测定回収的水溶液中的葡聚糖浓度,将各时刻的葡聚糖浓度除以试验结束后的葡聚糖浓度而得的值以百分数表示,将所得值作为透过率。
将结果示于图5。
将海藻酸衍生物(al-ex-2―1)/海藻酸衍生物(al-ex-7-1-2)交联而得到的交联海藻酸结构体、将海藻酸衍生物(al-ex-9)/海藻酸衍生物(al-ex-7-1-2)交联而得到的交联海藻酸结构体、将海藻酸衍生物(al-ex-8)/海藻酸衍生物(al-ex-7-1-2)交联而得到的交联海藻酸结构体、和将海藻酸衍生物(al-ex-10)/海藻酸衍生物(al-ex-7-1-2)交联而得到的交联海藻酸结构体均在3小时后显示约不到20%的泄漏率、在24小时显示约50%的泄漏率。
- 还没有人留言评论。精彩留言会获得点赞!