一种粉末尼龙制备装置、制备方法及粉末尼龙与流程
[0001]
本发明涉及尼龙材料技术领域,具体涉及一种粉末尼龙制备装置、制备方法及粉末尼龙。
背景技术:
[0002]
粉末尼龙是一种很有发展前景的高分子材料,其保持了原有尼龙优异的物理化学性能,具有较大的比表面积、多孔性结构及良好的加工性能,这使得粉末尼龙在各个领域得到广泛应用,现在应用较多的领域有粉末涂料、生物医药、胶黏剂、改性填料、化妆品等。
[0003]
专利cn104191615a公开了一种3d打印用高分子聚合物粉末材料的制备方法,具体为一种尼龙覆膜高分子聚合物粉末材料的制备方法,其选用强极性的溶剂将尼龙进行溶解,结晶析出,经真空干燥、球磨,筛分选择一定粒径分布的粉末即为尼龙覆膜高分子聚合物粉末材料,该制备工序存在强极性溶剂的回收以及环境污染问题。
[0004]
专利cn106893314a公开了一种耐磨损聚酰亚胺树脂/尼龙复合材料及其制备方法和应用,其中复合材料包括如下组分及其质量份数:尼龙粉末80-95份、聚酰亚胺树脂粉末0.5-20份、表面改性剂0.1-2份、流动助剂0.5-2份、抗氧化剂0.1-1份,经在氮气保护下的高压反应釜内融化、混匀、沉淀析出、过滤、清除溶剂、筛分等制备过程,成品粒径分布均匀,粒径范围在40-60μm之间、形态规整、流动性好,能作为3d打印材料,满足3d打印的性能要求。
[0005]
专利cn1473877a公开了尼龙1212盐、抗氧剂和分子量调节剂聚合得到尼龙1212树脂,再将尼龙1212树脂、颜料和助剂经双螺杆挤出造粒,深冷粉碎、整形和过筛等工序得到尼龙1212粉末,粉末尼龙的制备工序较为繁琐,生产效率较低。
[0006]
由此可见,现有的粉末尼龙的制备装置及制备工艺存在有机溶剂回收成本高、污染较大、工艺步骤较多和生产效率较低等问题。
技术实现要素:
[0007]
本发明要解决的技术问题是克服现有技术存在的不足,提供一种粉末尼龙制备装置和制备方法,可合成出分子量高、分子量分布宽度较窄、粒径均一的脂肪族粉末尼龙,其普遍应用于旋转成型、粉末涂料、压制烧结、3d打印等工业领域。
[0008]
为解决上述技术问题,本发明提出的技术方案为:
[0009]
一种粉末尼龙制备装置,包括预聚釜和出料增粘一体化装置,所述出料增粘一体化装置的输入端与所述预聚釜的输出端相连,所述出料增粘一体化装置包括装置本体和用于对所述装置本体内的物料进行破碎搅拌的破碎搅拌装置。出料增粘一体化装置与预聚釜配合能够得到分子量高、分子量分布宽度较窄、粒径均一的脂肪族粉末尼龙。
[0010]
上述的粉末尼龙制备装置,优选的,所述出料增粘一体化装置还包括换热夹套和用于控制所述装置本体内温度的模温机,所述换热夹套设于装置本体的外层。换热夹套和模温机可以对温度进行调控,压力喷料装置、破碎搅拌装置和模温机可实现在较低温度下得到粒径较小的粉末脂肪族尼龙,因此可较好地调控粉末尼龙的树脂粘度,从而满足不同
的性能要求。
[0011]
上述的粉末尼龙制备装置,优选的,所述出料增粘一体化装置还包括用于调节所述装置本体内压力的真空机组,且装置本体的体积为所述预聚釜体积的3~5倍,以便于进行后续的破碎成粉末尼龙及固相增粘处理。模温机、真空机组和固相增粘装置可实现一定分子量的低粘度脂肪族尼龙树脂的增粘,从而满足作为3d打印材料对于其力学强度的性能要求。
[0012]
上述的粉末尼龙制备装置,优选的,所述出料增粘一体化装置还包括压力喷料装置,所述压力喷料装置设于所述预聚釜和出料增粘一体化装置之间。压力喷料装置和破碎搅拌装置同时作用能够保障粉末尼龙的粉体粒径较为均一。
[0013]
作为一个总的技术构思,本发明还提供了一种粉末尼龙制备方法,采用上述的粉末尼龙制备装置,包括以下步骤,
[0014]
s1、将原料投入预聚釜中,并加入光稳定剂、分子量调节剂、次磷酸钠和去离子水;
[0015]
s2、用氮气置换预聚釜内的空气,然后升高预聚釜内的温度,并进行保压;
[0016]
s3、将预聚釜中的气体放出至常压,然后将预聚釜内抽真空进一步减压,得到尼龙树脂;
[0017]
s4、将尼龙树脂喷出至出料增粘一体化装置中进行破碎搅拌处理得到粉末尼龙。
[0018]
该制备方法工序简单,且没有使用有机溶剂,能够得到分子量高、分子量分布宽度窄、粒径均一的粉末尼龙,其中预聚先得到一定分子量的低粘度脂肪族尼龙树脂,再经过出料增粘一体化装置中的喷料、破碎等处理得到粒径较小的粉末脂肪族尼龙,过筛得到所需粒径的粉末脂肪族尼龙。
[0019]
上述的粉末尼龙制备方法,优选的,所述s1中原料为内酰胺、脂肪族尼龙盐中的一种或多种。
[0020]
上述的粉末尼龙制备方法,优选的,所述s2中先将预聚釜内的温度升至180℃~220℃,并保持预聚釜内的压力为1.5~2.5mpa,再将预聚釜内的温度升至200℃~260℃,并保持预聚釜内的压力为1.5~2.0mpa。
[0021]
上述的粉末尼龙制备方法,优选的,所述s3中将预聚釜中的气体放出至常压前保压1.5h,然后将预聚釜内抽真空进一步减压至-0.06mpa。
[0022]
上述的粉末尼龙制备方法,优选的,所述s4中通过压力喷料装置将尼龙树脂喷送至装置本体中,同时采用破碎搅拌装置对喷出的尼龙树脂进行破碎搅拌,通过换热夹套和模温机控制装置本体内的温度,通过真空机组调节装置本体的压力,对装置本体内的物料进行固相增粘处理,在一定温度和转速下,连续抽真空,带走体系中生产的水分子,使反应向正方向进行,从而逐步提升粉末尼龙的分子量。压力喷料装置和破碎搅拌装置保障粉末尼龙的粉体粒径较为均一,而固相增粘装置可较好地调控粉末尼龙的树脂粘度,从而满足不同的性能要求。
[0023]
作为一个总的技术构思,本发明还提供了一种采用上述的粉末尼龙制备装置或上述的粉末尼龙制备方法制备得到的粉末尼龙。
[0024]
上述的粉末尼龙,优选的,所述粉末尼龙为尼龙1010、尼龙6、尼龙66、尼龙1l、尼龙12、二元共聚尼龙、三元共聚尼龙中的一种或多种。
[0025]
与现有技术相比,本发明的优点在于:
[0026]
1.本发明的粉末尼龙制备装置将预聚釜和出料增粘一体化装置组成一体,由预聚釜得到的尼龙树脂在出料增粘一体化装置中被喷出,同时被高速破碎并在一定温度和压力条件下进行固相增粘处理,处理后可得到分子量高、分子量分布宽度较窄、粒径均一的脂肪族粉末尼龙,普遍应用于旋转成型、粉末涂料、压制烧结、3d打印等工业领域。
[0027]
2.本发明的粉末尼龙制备方法采用内酰胺、脂肪族尼龙盐为原材料,经均聚或共聚合成具有一定分子量的尼龙树脂,然后通过出料增粘一体化装置进行压力自动化喷料、高速破碎和固相增粘处理,工序简单、整个聚合生产过程不使用有机溶剂,只采用去离子水作为反应溶剂,绿色环保,可重复使用,实现零排放。
[0028]
3.本发明制备得到的粉末尼龙具有分子量高、分子量分布宽度较窄等优点,该粉末尼龙和无机纳米颗粒的结合为合成具有优越的机械、电学、光学或磁性能的复合微球提供了机会。
附图说明
[0029]
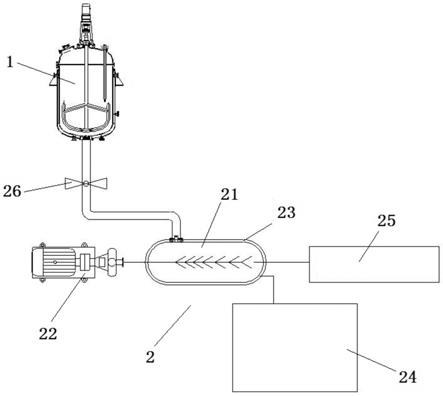
图1是实施例的粉末尼龙制备装置的结构示意图。
[0030]
图例说明:
[0031]
1、预聚釜;2、出料增粘一体化装置;21、装置本体;22、破碎搅拌装置;23、换热夹套;24、模温机;25、真空机组;26、压力喷料装置。
具体实施方式
[0032]
为了便于理解本发明,下文将结合说明书附图和较佳的实施例对本发明做更全面、细致地描述,但本发明的保护范围并不限于以下具体实施例。
[0033]
实施例1:
[0034]
将5kg己内酰胺投入预聚釜1中,并加入4g光稳定剂tinuvin 622、10g分子量调节剂、50g次磷酸钠和500g去离子水,用高纯氮气置换预聚釜1内空气3~4次,升温至180℃,保持釜内压力1.5~2.5mpa,继续升温至230℃,保持釜内压力1.5~2.0mpa,保压1.5h后放气至常压,逐步抽真空,使体系减压至-0.06mpa,即得到具有一定分子量的尼龙树脂,经压力喷料装置26、破碎搅拌装置22及固相增粘处理后可得到分子量高、分子量分布宽度较窄、粒径均一的脂肪族粉末尼龙,测其相对粘度为2.45,分子量分布宽度为1.54,粒径为0.1mm。
[0035]
实施例2:
[0036]
将5kg十二内酰胺投入预聚釜1中,并加入4g光稳定剂tinuvin 622、10g分子量调节剂、50g次磷酸钠和500g去离子水,用高纯氮气置换预聚釜1内空气3~4次,升温至180℃,保持釜内压力1.5~2.5mpa,继续升温至200℃,保持釜内压力1.5~2.0mpa,保压1.5h后放气至常压,逐步抽真空,使体系减压至-0.06mpa,即得到具有一定分子量的尼龙树脂,经压力喷料装置26、破碎搅拌装置22及固相增粘处理后可得到分子量高、分子量分布宽度较窄、粒径均一的脂肪族粉末尼龙,测其相对粘度为2.48,分子量分布宽度为1.50,粒径为0.1mm。
[0037]
实施例3:
[0038]
将5kg氨基十一酸投入预聚釜1中,并加入4g光稳定剂tinuvin 622、10g分子量调节剂、50g次磷酸钠和500g去离子水,用高纯氮气置换预聚釜1内空气3~4次,升温至180℃,保持釜内压力1.5~2.5mpa,继续升温至210℃,保持釜内压力1.5~2.0mpa,保压1.5h后放
气至常压,逐步抽真空,使体系减压至-0.06mpa,即得到具有一定分子量的尼龙树脂,经压力喷料装置26、破碎搅拌装置22及固相增粘处理后可得到分子量高、分子量分布宽度较窄、粒径均一的脂肪族粉末尼龙,测其相对粘度为2.46,分子量分布宽度为1.48,粒径为0.1mm。
[0039]
实施例4:
[0040]
将5kg尼龙66盐投入预聚釜1中,并加入4g光稳定剂tinuvin 622、10g分子量调节剂、50g次磷酸钠和500g去离子水,用高纯氮气置换预聚釜1内空气3~4次,升温至220℃,保持釜内压力1.5~2.5mpa,继续升温至260℃,保持釜内压力1.5~2.0mpa,保压1.5h后放气至常压,逐步抽真空,使体系减压至-0.06mpa,即得到具有一定分子量的尼龙树脂,经压力喷料装置26、破碎搅拌装置22及固相增粘处理后可得到分子量高、分子量分布宽度较窄、粒径均一的脂肪族粉末尼龙,测其相对粘度为2.50,分子量分布宽度为1.55,粒径为0.1mm。
[0041]
实施例5:
[0042]
将5kg尼龙1010盐投入预聚釜1中,并加入4g光稳定剂tinuvin 622、10g分子量调节剂、50g次磷酸钠和500g去离子水,用高纯氮气置换预聚釜1内空气3~4次,升温至180℃,保持釜内压力1.5~2.5mpa,继续升温至220℃,保持釜内压力1.5~2.0mpa,保压1.5h后放气至常压,逐步抽真空,使体系减压至-0.06mpa,即得到具有一定分子量的尼龙树脂,经压力喷料装置26、破碎搅拌装置22及固相增粘处理后可得到分子量高、分子量分布宽度较窄、粒径均一的脂肪族粉末尼龙,测其相对粘度为2.48,分子量分布宽度为1.58,粒径为0.1mm。
[0043]
实施例6:
[0044]
将2.5kg己内酰胺和2.5kg尼龙66盐投入预聚釜1中,并加入4g光稳定剂tinuvin 622、10g分子量调节剂、50g次磷酸钠和500g去离子水,用高纯氮气置换预聚釜1内空气3~4次,升温至180℃,保持釜内压力1.5~2.5mpa,继续升温至240℃,保持釜内压力1.5~2.0mpa,保压1.5h后放气至常压,逐步抽真空,使体系减压至-0.06mpa,即得到具有一定分子量的尼龙树脂,经压力喷料装置26、破碎搅拌装置22及固相增粘处理后可得到分子量高、分子量分布宽度较窄、粒径均一的脂肪族粉末尼龙,测其相对粘度为2.48,分子量分布宽度为1.51,粒径为0.1mm。
[0045]
实施例7:
[0046]
将2.5kg己内酰胺和2.5kg尼龙1010盐投入预聚釜1中,并加入4g光稳定剂tinuvin622、10g分子量调节剂、50g次磷酸钠和500g去离子水,用高纯氮气置换预聚釜1内空气3~4次,升温至180℃,保持釜内压力1.5~2.5mpa,继续升温至220℃,保持釜内压力1.5~2.0mpa,保压1.5h后放气至常压,逐步抽真空,使体系减压至-0.06mpa,即得到具有一定分子量的尼龙树脂,经压力喷料装置26、破碎搅拌装置22及固相增粘处理后可得到分子量高、分子量分布宽度较窄、粒径均一的脂肪族粉末尼龙,测其相对粘度为2.52,分子量分布宽度为1.52,粒径为0.1mm。
[0047]
实施例8:
[0048]
将2.5kg尼龙66和2.5kg尼龙1010盐投入预聚釜1中,并加入4g光稳定剂tinuvin622、10g分子量调节剂、50g次磷酸钠和500g去离子水,用高纯氮气置换预聚釜1内空气3~4次,升温至180℃,保持釜内压力1.5~2.5mpa,继续升温至230℃,保持釜内压力1.5~2.0mpa,保压1.5h后放气至常压,逐步抽真空,使体系减压至-0.06mpa,即得到具有一定分子量的尼龙树脂,经压力喷料装置26、破碎搅拌装置22及固相增粘处理后可得到分子
量高、分子量分布宽度较窄、粒径均一的脂肪族粉末尼龙,测其相对粘度为2.49,分子量分布宽度为1.56,粒径为0.1mm。
[0049]
上述全部实施例均采用本实施例的粉末尼龙制备装置进行制备。
[0050]
如图1所示,本实施例的粉末尼龙制备装置,包括预聚釜1和出料增粘一体化装置2,出料增粘一体化装置2的输入端与预聚釜1的输出端相连,出料增粘一体化装置2包括装置本体21和用于对装置本体21内的物料进行破碎搅拌的破碎搅拌装置22。
[0051]
本实施例中,出料增粘一体化装置2还包括换热夹套23和用于控制装置本体21内温度的模温机24,换热夹套23设于装置本体21的外层。
[0052]
本实施例中,出料增粘一体化装置2还包括用于调节装置本体21内压力的真空机组25,且装置本体21的体积为预聚釜1体积的3~5倍。
[0053]
本实施例中,出料增粘一体化装置2还包括压力喷料装置26,压力喷料装置26设于预聚釜1和出料增粘一体化装置2之间。具体的,本实施例的压力喷料装置26采用自动球阀。
[0054]
下表1示出了实施例1至实施例8制备得到的粉末尼龙的相关性能数据。
[0055]
表1实施例粉末尼龙的相关性能数据
[0056][0057]
上述各性能数据的测试方法与条件如下:
[0058]
熔点测试条件:称取样品5~8mg,氮气保护下,样品升温至270℃熔融3min,用液氮淬冷,然后将淬冷的样品升温至350℃,降至常温,再升温至350℃,升温速率均为10℃/min。
[0059]
初始分解温度测试条件:称取样品5~8mg,氮气保护下,样品以10℃/min升温至800℃,样品失重5%wt对应的温度。
[0060]
拉伸强度测试条件:将拉伸样条置于恒温恒湿箱中处理24h,使用试验机进行测试,测试标准为gb/t 1040.2-2006。
[0061]
弯曲强度测试条件:将弯曲样条置于恒温恒湿箱中处理24h,使用试验机进行测试,测试标准为gb/t 9341-2008。
[0062]
缺口冲击强度测试条件:将冲击样条置于恒温恒湿箱中处理24h,使用试验机进行测试,测试标准为gb/t 1043.1-2008。
[0063]
虽然本发明已以较佳实施例揭露如上,然而并非用以限定本发明。任何熟悉本领域的技术人员,在不脱离本发明技术方案范围的情况下,都可利用上述揭示的技术内容对本发明技术方案做出许多可能的变动和修饰,或修改为等同变化的等效实施例。因此,凡是未脱离本发明技术方案的内容,依据本发明技术实质对以上实施例所做的任何简单修改、
等同变化及修饰,均应落在本发明技术方案保护的范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1