生物催化制备(S)-3-丁炔-2-丙炔胺的方法与流程
本发明涉及生物催化化学反应领域,特别是涉及一种生物催化制备对映体纯的(s)-3-丁炔-2-丙炔胺的方法。
背景技术:
(s)-3-丁炔-2-丙炔胺(如式ii所示),是一种非常重要的手性砌块,应用于很多医药中间体的合成。例如(s)-3-丁炔-2-丙炔胺是抑制神经退行性疾病药物的制备路线中重要的药物中间体。
目前,制备(s)-3-丁炔-2-丙炔胺的方法如下图示,文献(chin.j.chem.2013,31,173–181)中以(r)-(+)-3-丁炔-2-醇为原料,并且使用三苯基磷和联氨这些剧毒不稳定的原料经过一步反转反应和一步脱去保护基团的反应,得到产品(s)-3-丁炔-2-丙炔胺。但是作为原材料的(r)-(+)-3-丁炔-2-醇价格昂贵,从经济角度出发不适合工业化大规模生产。
而生物酶催化法因为具有反应效率高、立体选择性好、反应条件温和、低能耗、环境友好等优点,受到越来越广泛的关注和越来越深入的研究。
目前关于(s)-3-丁炔-2-丙炔胺的生物制备方法仍没有相关专利报道。
技术实现要素:
本发明所要解决的技术问题在于,提供一种高产率、选择性好、路线简化的(s)-3-丁炔-2-丙炔胺的制备方法,主要解决现有路线步骤中过渡金属催化烯胺或亚胺加氢的步骤较为繁琐,转化率不高或选择性不高的技术问题。
为解决上述技术问题,本发明采用的技术方案为
一种生物催化制备(s)-3-丁炔-2-丙炔胺的方法,在液相反应体系中,以3-炔基-2-丁酮为底物,在辅酶存在的条件下,反应体系的ph为8.0~9.0,反应温度为20℃~40℃,使用亚胺还原酶催化将3-炔基-2-丁酮的羰基还原为手性氨基制备得到(s)-3-丁炔-2-丙炔胺。
反应式如下:
在一种优选实施例中,所述亚胺还原酶选自以下组:
(1)亚胺还原酶aoired,其氨基酸序列如seqidno:1所示;
(2)以及对seqidno:1所示的氨基酸序列在保持酶活性范围内,进行一个或多个氨基酸的替换、缺失、改变、插入或增加,所得到的氨基酸序列。
在一种优选实施例中,所述亚胺还原酶的编码基因序列选自以下组:
(a)如seqidno:2所示的核苷酸序列;
(b)与(a)限定的序列互补的多核苷酸;或
(c)与(a)限定的序列具有至少70%(优选至少75%、80%、85%、90%,更优选至少95%、96%、97%、98%、99%)以上的序列一致性的任一多核苷酸或互补序列。
在一种优选实施例中,所述亚胺还原酶的编码基因工件在表达载体上。
在本发明中,所述亚胺还原酶的来源包括但不限于弧菌、节杆菌和色杆菌。其中优选的转氨酶来源于东方拟无枝酸菌(uniprotr4snk4)中发现的新型亚胺还原酶(aoired)或来源于将所述菌种的基因重组于大肠杆菌中。
在一种优选实施例中,所述反应体系中,所述3-炔基-2-丁酮的反应起始浓度为1g/l-5g/l。
在一种优选实施例中,所述亚胺还原酶的用量为底物质量的1-3倍。
在一种优选实施例中,所述的辅酶选自:还原性辅酶,氧化性辅酶,或其组合。
在一种优选实施例中,所述还原性辅酶选自nadh、nadph、或其组合。
在一种优选实施例中,所述氧化性辅酶选自nad、nadp、或其组合。较佳地,所述的辅酶选自还原性辅酶nadp。
在一种优选实施例中,所述辅酶用量与底物用量比率为0.01%~1.0%(w/w)。较佳地,辅酶用量与底物用量比率为0.01%~0.5%(w/w)。
在一种优选实施例中,所述反应体系中,还存在共底物。
在一种优选实施例中,所述共底物选自:异丙醇、葡萄糖、甲酸铵中的一种或多种。较佳地,所述共底物选自葡萄糖。
在一种优选实施例中,所述反应体系中,共底物的浓度为5-30%。
在一种优选实施例中,所述反应体系中,还存在用于辅酶再生的酶。
在一种优选实施例中,所述用于辅酶再生的酶选自:醇脱氢酶、甲酸脱氢酶、葡萄糖脱氢酶中的一种或多种。较佳地,所述用于辅酶再生的酶选自葡萄糖脱氢酶(gdh)。
在一种优选实施例中,所述反应体系中,所述用于辅酶再生的酶的用量为辅酶质量的1%-10%。
在一种优选实施例中,所述的反应体系为缓冲盐水溶液体系。通过缓冲液来对反应体系的ph进行控制。常用的缓冲液包括但不限于氯化铵(nh4cl)、甲酸胺(hcoonh4)或醋酸铵(ch3coonh4)缓冲液。
在一种优选实施例中,所述的反应体系还含有助溶剂。所述助溶剂为有机溶剂。
在一种优选实施例中,所述的助溶剂选自下组:二甲基亚砜,甲醇,乙醇,异丙醇,丙酮,或其组合。较佳地,所述助溶剂选自二甲基亚砜。所使用的助溶剂应当与水可混溶,以进一步增加底物的溶解性。
在一种优选实施例中,反应温度为25℃~35℃;更佳地30℃~35℃。
在一种优选实施例中,反应时间为0.1~120小时;较佳地0.5~48小时;更佳地1~24小时。
在一种优选实施例中,较佳地,反应体系的ph为8.5±0.3;更佳地,ph为8.5±0.2;更佳地,ph为8.5±0.1;更佳地,ph为8.5。
在一种优选实施例中,本发明的制备方法还包括分离出羟基产物(s)-3-丁炔-2-丙炔胺的步骤。
在优选的条件下,以本发明的方法制备的(s)-3-丁炔-2-丙炔胺(ii),对映体过量值不低于99%,转化率不低于81%。更优选的条件下,转化率不低于99%。
本发明通过一种新型酶催化技术,实现制备对映体纯的(s)-3-丁炔-2-丙炔胺的目的。该方法从3-炔基-2-丁酮出发,使用筛选的特定氨基酸序列的亚胺还原酶来催化反应(s)-3-丁炔-2-丙炔胺,在反应中还可以加入还原型辅nadph来作为电子传递,还可以配合使用葡萄糖脱氢酶实行辅酶循环。该条技术路线步骤简单,转化率高,技术绿色环保,具有工业化生产的潜力。
本发明的有益效果是,从3-炔基-2-丁酮出发,使用新型亚胺还原酶来催化反应s-3-丁炔-2-丙炔胺的技术路线在操作步骤上简单易行、产率高、选择性好,对底物的转化率高达81%以上(更优选的条件下达99%以上),所获得产品的手性纯度达99%,合成路线简洁高效,反应条件温和易控,反应时间短,绿色环保,为工业化生产(s)-3-丁炔-2-丙炔胺提供了新的思路与机会,具有工业化推广的潜力。
附图说明
图1是3-丁炔-2-丙炔胺的外消旋物的高效液相色谱法图谱。左侧的峰为r构型,右侧的峰为s构型。
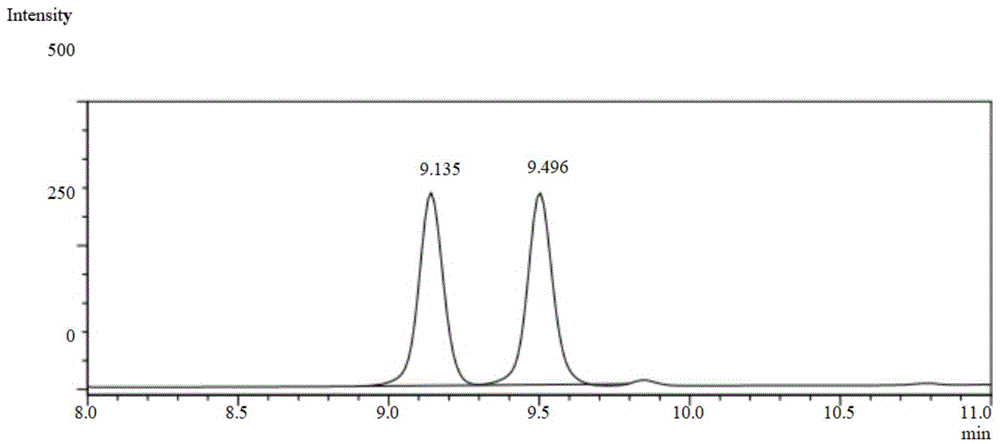
图2a是按照本发明实施例1的方法转化后的s-3-丁炔-2-丙炔胺的手性高效液相色谱法图谱。图中唯一的峰为目标化合物s-3-丁炔-2-丙炔胺。
图2b是按照本发明实施例1的方法(氯化铵ph值为8.5)转化后的(s)-3-丁炔-2-丙炔胺的转化率气象色谱法图谱。图中唯一的峰为目标化合物s-3-丁炔-2-丙炔胺。
图3是按照本发明实施例2的方法(氯化铵ph值为8.5)转化后的(s)-3-丁炔-2-丙炔胺的转化率气象色谱法图谱。
图4是按照本发明实施例3的方法(氯化铵ph值为10.0)转化后的(s)-3-丁炔-2-丙炔胺的转化率气象色谱法图谱。
图5是按照本发明实施例4的方法(氯化铵ph值为6.0)转化后的(s)-3-丁炔-2-丙炔胺的转化率气象色谱法图谱。
图6是按照本发明实施例5的方法(氯化铵ph值为7.0)转化后的(s)-3-丁炔-2-丙炔胺的转化率气象色谱法图谱。
图7是按照本发明实施例6的方法(氯化铵ph值为9.5)转化后的(s)-3-丁炔-2-丙炔胺的转化率气象色谱法图谱。
具体实施方式
下面将对本发明的技术方案进行清楚、完整的描述,显然,所描述的实施例是本发明的一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动的前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例1
在8ml玻璃反应瓶中,加入4ml氯化铵缓冲液。少量多次加入氨水,用玻璃棒持续搅拌,在反应液达到均匀后,将电极没入反应液,再次测量反应液ph值,调节ph值到8.5后轻摇玻璃反应瓶,电极没入反应液后等待数字稳定后读取,平行测量三次,并记录,最后取三次测量值的平均值,反应液ph值为8.5。测量结束后用蒸馏水清洗电极,用滤纸将蒸馏水吸干后,套上复合电极套,该测量步骤完成。然后向反应液中加入50mg亚胺还原酶冻干粉和1mg烟酰胺腺嘌呤二核苷酸磷酸(氧化态)(nadp),5mg葡萄糖脱氢酶(gdh)和100mg葡萄糖搅拌充分后加入20mg底物,放入全温度恒温培养摇床调整并控制反应温度为35℃,连续搅拌24小时,使其充分反应。
待反应结束后,取1ml反应液加入1ml乙腈,滴入1滴苯甲酰氯,放入摇床振荡15分钟后放入离心机离心处理,分层后,取上层所需层,即为粗产品的溶液。将粗产品溶液通入氮气吹干除去杂质后,加入1ml乙醇,进入高效液相色谱仪进行分析,检测反应选择性情况。分析条件:岛津lc-20a液相色谱仪和紫外检测器,chiralpakod-3r,150mm×4.6mm,3μm手性色谱柱,流动相:含0.05%三氟乙酸的乙腈-含0.05%三氟乙酸的水。30℃1ml/min下平衡,检测波长220nm。
由于产品没有紫外吸收,因此采用衍生化方法结合液相色谱来检测产品的手性纯度。产品的液相色谱法手性图谱见图2a。图2a显示,t=9.5处唯一的峰为目标化合物(s)-3-丁炔-2-丙炔胺。反应完成后,经处理得粗产品的溶液经检测,所获得产品的手性纯度大于99%。
此外,取1ml反应液加入1ml二氯甲烷萃取,萃取结束后,萃取混合物放入离心机离心处理,分层后,取下层所需层,即为粗产品的溶液。进入高效气相色谱仪进行分析,检测反应纯度情况。分析条件:岛津gc-2010plus气象色谱仪,带fid检测器,db-ffap(30m*0.32mmid*1.0μm)或等效柱,h2:40ml/min,air:400ml/min,进样口温度:180℃,进样分流比:50:1,柱流速:30.0cm/sec,恒线速度模式。
图2b为汽相色谱图谱,用于确认反应的转化率。图2b显示,t=5.4处唯一的峰为目标化合物(s)-3-丁炔-2-丙炔胺,转化率达99%以上。
实施例2
首先在1l的纯水中加入21.40g氯化铵,充分搅拌至固体澄清,配制成400mmol的氯化铵缓冲液。用ph计测量该缓冲液浓度,初始ph值为7.5。在8ml玻璃反应瓶中,加入4ml氯化铵缓冲液。加入氨水调节ph值,使8ml玻璃反应瓶中的反应液达到8.5。然后向反应液中加入50mg亚胺还原酶(aoired)冻干粉和1mg烟酰胺腺嘌呤二核苷酸磷酸(氧化态)(nadp),5mg葡萄糖脱氢酶(gdh),和100mg葡萄糖搅拌充分后加入20mg底物炔酮。放入全温度恒温培养摇床,调整并控制反应温度30℃,连续恒温搅拌24小时,使其充分反应。
取1ml反应液加入1ml二氯甲烷萃取,萃取结束后,萃取混合物放入离心机离心处理,分层后,取下层所需层,即为粗产品的溶液。进入高效气相色谱仪进行分析,检测反应纯度情况。分析条件:岛津gc-2010plus气象色谱仪,带fid检测器,db-ffap(30m*0.32mmid*1.0μm)或等效柱,h2:40ml/min,air:400ml/min,进样口温度:180℃,进样分流比:50:1,柱流速:30.0cm/sec,恒线速度模式。
采用汽相色谱来确认反应的转化率。产品的汽相色谱法手性图谱见图3。图3显示,t=5.1处的出峰为原料,t=5.5处出峰为标化合物(s)-3-丁炔-2-丙炔胺,转化率达81%以上。
实施例3
首先在1l的纯水中加入21.40g氯化铵,充分搅拌至固体澄清,配制成400mmol的氯化铵缓冲液。用ph计测量该缓冲液浓度,初始ph值为7.5。在8ml玻璃反应瓶中,加入4ml氯化铵缓冲液。加入氨水调节ph值,使8ml玻璃反应瓶中的反应液达到10。然后向反应液中加入50mg亚胺还原酶(aoired)冻干粉和1mg烟酰胺腺嘌呤二核苷酸磷酸(氧化态)(nadp),5mg葡萄糖脱氢酶(gdh),和100mg葡萄糖搅拌充分后加入20mg底物炔酮。放入全温度恒温培养摇床,调整并控制反应温度30℃,连续恒温搅拌24小时,使其充分反应。
取1ml反应液加入1ml二氯甲烷萃取,萃取结束后,萃取混合物放入离心机离心处理,分层后,取下层所需层,即为粗产品的溶液。进入高效气相色谱仪进行分析,检测反应纯度情况。分析条件:岛津gc-2010plus气象色谱仪,带fid检测器,db-ffap(30m*0.32mmid*1.0μm)或等效柱,h2:40ml/min,air:400ml/min,进样口温度:180℃,进样分流比:50:1,柱流速:30.0cm/sec,恒线速度模式。
产品的汽相色谱法手性图谱见图4。图4显示,t=5.1处出峰为原料,t=5.571处出峰为标化合物(s)-3-丁炔-2-丙炔胺,转化率仅4%。可见,反应液ph值达到10时,不利于反应的进行。
对比例:
按照实施例2-3,在其它反应条件相同的前提下考察不同ph对反应转化率的影响,实验结果如下所示:
如图5~7所示,在ph6.0和7.0时,不利于反应的进行;在ph9.5时,反应的转化率也不高。
综上所述,上述各实施例仅为本发明的较佳实施例而已,并不用以限定本发明的保护范围,凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,皆应包含在本发明的保护范围内。
序列表
<110>上海合全药物研发有限公司
<120>生物催化制备(s)-3-丁炔-2-丙炔胺的方法
<130>cpc-np-20-101842
<160>2
<170>siposequencelisting1.0
<210>1
<211>290
<212>prt
<213>人工序列(artificialsequence)
<400>1
metthraspglnasnleuprovalthrvalalaglyleuglypromet
151015
glyseralaleualaalaalaleuleuaspargglyhisaspvalthr
202530
valtrpasnargserproglylysalaalaproleuvalalalysgly
354045
alaargglnalaaspaspilevalaspalavalseralaserargleu
505560
leuvalvalcysleualaasptyraspalaleutyrseralaleugly
65707580
proalaargglualaleuargglyargvalvalvalasnleuasnser
859095
glythrprolysglualaasnglualaleuargtrpalagluarghis
100105110
glythrglytyrleuaspglyalailemetvalproproalametval
115120125
glyhisproglyservalpheleutyrserglyseralagluvalphe
130135140
gluglutyrlysgluthrleualaglyleuglyaspprovalhisleu
145150155160
glythrglualaglyleualavalleutyrasnthralaleuleuser
165170175
metmettyrsersermetasnglypheleuhisalaalaalaleuval
180185190
glyseralaglyvalproalaalagluphethrlysleualavalasp
195200205
trppheleuproalavalileglyglnileilelysalaglualapro
210215220
thrileaspgluglyvaltyrproglyaspalaglyserleuglumet
225230235240
asnvalthrthrleulyshisileileglythrserglngluglngly
245250255
valaspthrgluileprovalargasnlysgluleuleuaspargala
260265270
valalaalaglypheglyglusersertyrtyrservalilegluleu
275280285
trparg
290
<210>2
<211>873
<212>dna
<213>人工序列(artificialsequence)
<400>2
atgaccgatcagaacctcccggtgaccgtcgccgggctcggcccgatgggctccgcgctc60
gccgccgccctgctcgaccgcggtcacgacgtgaccgtgtggaaccgctcccccggcaag120
gccgcccccttggtggcgaaaggcgcgcggcaggcggacgacatcgtggacgcggtgtcc180
gcgagccgtctgctcgtggtctgcctcgccgactacgacgcgctttactccgcgctcggc240
cccgcgcgggaagccttgcgcggccgcgtagtggtgaacctgaattccggcacgccgaag300
gaggcgaacgaagctctccgatgggccgagcgacacggaacgggctatctcgacggcgcc360
atcatggttccccccgcgatggtcggccaccccggctcggtcttcctctacagcggttcc420
gccgaggttttcgaggaatacaaggagacattggccggtctgggtgatccggtccatctc480
ggcacggaagccggcctcgccgtgctgtacaacaccgcgttgctgagcatgatgtactcg540
tcgatgaacggtttcctccacgccgccgcgctggtcggcagtgccggggtcccggcggcc600
gaattcacgaagctcgccgtcgactggttcctgcccgcggtgatcggacagatcatcaag660
gcggaggcgcccaccatcgacgaaggcgtgtaccccggtgacgccggttcgctggaaatg720
aacgtcacgacactgaagcacatcatcggaaccagccaggagcagggcgtcgacaccgag780
atcccggtccgcaacaaggaacttctggaccgggccgtcgccgccgggttcggcgagagc840
agctattactcggtgatcgaactgtggaggtga873
- 还没有人留言评论。精彩留言会获得点赞!