一种基于透析生物膜的高质量真菌DNA提取方法
一种基于透析生物膜的高质量真菌dna提取方法
技术领域
1.本发明涉及dna提取技术领域,具体为一种基于透析生物膜的高质量真菌dna提取方法。
背景技术:
2.地球生物圈中存在着大量的微生物,其中真菌占据着十分重要的地位,据估计全世界有寄生和内生真菌至少250万种,而仅有不到97861种真菌被人类所认识和命名,仅占估计种数的3.9%,尚有96.1%的真菌有待人类去命名、描述,明确真菌种类对于研究生命科学、开发利用真菌资源具有重要意义。随着分子生物学的发展,核苷酸序列分析能够快速鉴定出已知种类,是真菌鉴定过程中的重要部分,其中dna的提取是核苷酸序列分析的基础,获取高浓度dna也是众多分子实验最为重要的一步,目前用于真菌基因组dna提取的方法有ctab法、sds法、氯化苄法、chelex-100法等。目前实验室里常用试剂盒法提取真菌dna,目前多种情况均会影响到dna的提取效率,比如真菌细胞壁的成分较多较为复杂的成分,如富含多糖、色素、核酸酶等物质;有些真菌在培养过程中产生大量的次生代谢产物等使得真菌dna提取比较困难。此外,在实验中,刮取真菌菌丝是提取dna过程中较为基础的一步,但真菌菌丝常与固体培养基里面的基质生长在一起,在实际操作过程中使得刮取有效菌丝十分困难,最终影响dna的浓度,导致测序结果欠佳,因此为提高dna的提取效率,解决这一问题十分必要。
3.塞洛芬(cellophane),通常称为“玻璃纸”,呈透明状,是一种用木材或者麻类以及其他植物的纤维制成的一种完全可降解的材料,类似一层塑料薄膜,实验室一般将其称为生物膜,不同于一般的塑料膜,该材料具有无毒、无味,安全环保等优点广泛运用于食品包装等领域,由于其材料的特殊性,微小的分子可以穿过这层薄膜,一些微生物也可在该材料上进行呼吸、生长,因此该生物膜开始在实验室用于真菌、细菌等微生物dna的提取,通常在固体培养基上贴上灭菌后的生物膜,将病原菌与培养基基质隔开,病原菌可透过生物膜正常生长,可有效解决菌丝生长在培养基基质里导致菌丝难以刮取最终影响dna浓度的问题。因此,与传统提取方法相比,生物膜的使用可大大提高dna的提取效率。
技术实现要素:
4.本发明的目的在于提供一种基于透析生物膜的高质量真菌dna提取方法,以解决上述背景技术中提出的问题。
5.为了解决上述技术问题,本发明提供如下技术方案:一种基于透析生物膜的高质量真菌dna提取方法,包括以下步骤:
6.步骤一:pda培养基的制备
7.称取pda培养基粉末于去离子水中并于高温灭菌锅中灭菌14-16min;
8.步骤二:病原菌接种
9.将pda平板设置贴膜处理,将灭菌后的pda培养基于超净工作台倒入培养皿中,待
培养基凝固后,在pda平板上用无菌镊子贴上彻底灭菌后的生物膜,然后在培养皿中心接种病原菌,将样品在26-30℃培养箱中恒温培养6-8d,待菌丝长满平板后用备用;
10.步骤三:真菌dna的提取及pcr扩增
11.利用真菌dna提取试剂盒进行dna的提取,采用真菌rdna-its通用引物对菌株进行pcr扩增。
12.在一种优选的实施方式中,所述步骤二中接种的病原菌预先进行纯化,病原菌的纯化方法为:将病原真菌无污染的边缘菌丝挑至pda平板上培养,重复此步骤直至获得纯培养菌株,将培养的菌株在26-30℃培养箱中恒温培养6-8d。
13.在一种优选的实施方式中,所述步骤三中dna提取试剂盒为biomiga fungal gdna kit试剂盒,所述步骤三中真菌rdna-its通用引物为its1:5
’‑
tccgtaggtgaacctgcgg-3’,its4:5
’‑
tcctccgcttattgatatgg-3’。
14.在一种优选的实施方式中,所述步骤三中对对菌株进行pcr扩增时,pcr扩增程序为94℃预变性3min,94℃变性30s,55℃退火45s,72℃延伸1min,共35个循环,反应总体系为25μl,包括2
×
taq pcr mix12.5μl、dna模板1μl、上下游引物各1μl,ddh2o 9.5μl。
15.与现有技术相比,本发明所达到的有益效果是:
16.本发明采用塞洛芬生物膜对dna进行提取,在相同的培养条件下可以大大提高dna的提取效率,而且对不同的真菌dna提取时没有显著性差异,贴生物膜处理的各真菌dna浓度显著高于未贴膜处理,其中燕麦镰刀菌贴膜的dna浓度是未贴膜的10倍之多,pcr产物扩增凝胶电泳图也表明贴膜样品的目的条带比未贴膜样品条带更为明显,此外该方法操作简便,易获得,成本较低,可广泛应用于实验室真菌dna的高效提取。
附图说明
17.附图用来提供对本发明的进一步理解,并且构成说明书的一部分,与本发明的实施例一起用于解释本发明,并不构成对本发明的限制。在附图中:
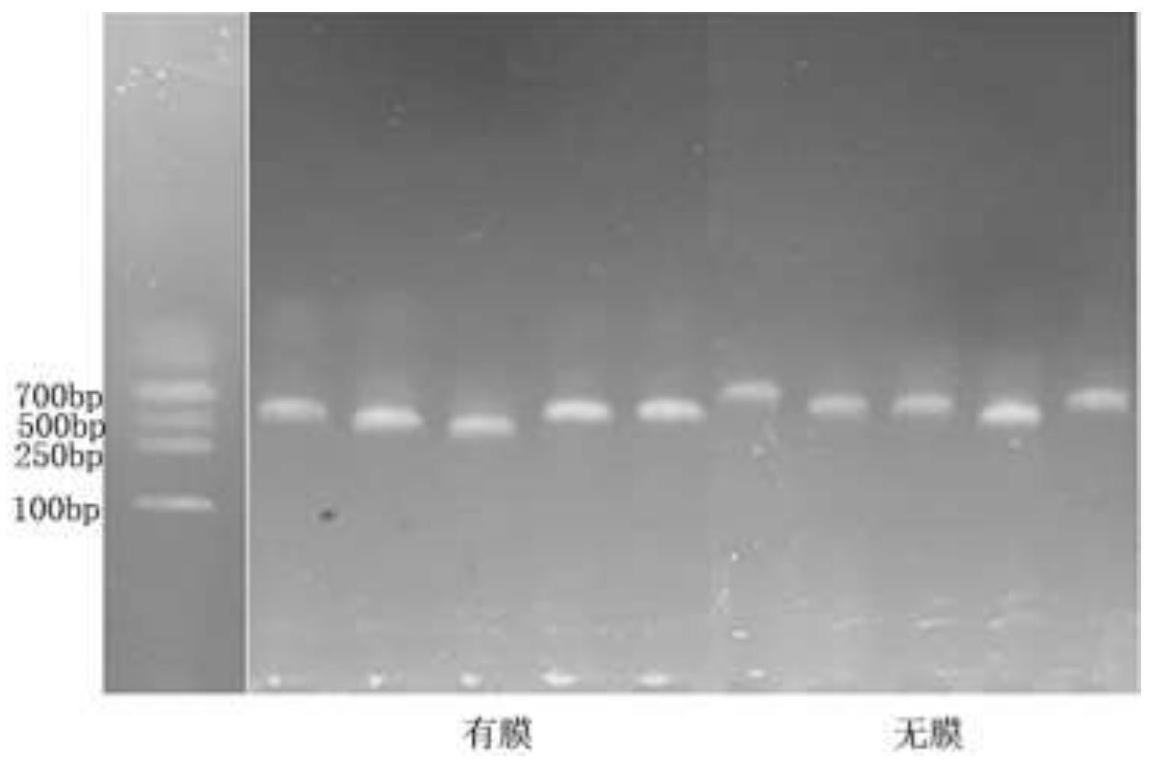
18.图1是本发明凝胶电泳示意图。
具体实施方式
19.下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
20.实施例1:
21.请参阅图1,本发明提供一种基于透析生物膜的高质量真菌dna提取方法,包括以下步骤:
22.步骤一:pda培养基的制备
23.称取46g pda培养基粉末于100ml去离子水中并于高温灭菌锅中灭菌15min;
24.步骤二:病原菌接种
25.将pda平板设置贴膜处理,将灭菌后的pda培养基于超净工作台倒入培养皿中,待培养基凝固后,在pda平板上用无菌镊子贴上彻底灭菌后的生物膜,然后在培养皿中心接种
立枯丝核菌病原菌,将样品在28℃培养箱中恒温培养7d,待菌丝长满平板后用备用;
26.步骤三:真菌dna的提取及pcr扩增
27.利用真菌dna提取试剂盒进行dna的提取,采用真菌rdna-its通用引物对菌株进行pcr扩增。
28.在一种优选的实施方式中,所述步骤二中接种的病原菌预先进行纯化,病原菌的纯化方法为:将病原真菌无污染的边缘菌丝挑至pda平板上培养,重复此步骤直至获得纯培养菌株,将培养的菌株在28℃培养箱中恒温培养7d。
29.在一种优选的实施方式中,所述步骤三中dna提取试剂盒为biomiga fungal gdna kit试剂盒,所述步骤三中真菌rdna-its通用引物为its1:5
’‑
tccgtaggtgaacctgcgg-3’,its4:5
’‑
tcctccgcttattgatatgg-3’。
30.在一种优选的实施方式中,所述步骤三中对对菌株进行pcr扩增时,pcr扩增程序为94℃预变性3min,94℃变性30s,55℃退火45s,72℃延伸1min,共35个循环,反应总体系为25μl,包括2
×
taq pcr mix12.5μl、dna模板1μl、上下游引物各1μl,ddh2o 9.5μl。
31.实施例2:
32.与实施例1不同的是,步骤二中接种的病原菌为尖孢镰刀菌。
33.实施例3:
34.与实施例1不同的是,步骤二中接种的病原菌为黑孢菌。
35.实施例4:
36.与实施例1不同的是,步骤二中接种的病原菌为绿色木酶。
37.实施例5:
38.与实施例1不同的是,步骤二中接种的病原菌为燕麦镰刀菌。
39.实施例6:
40.本发明提供一种真菌dna提取方法,包括以下步骤:
41.步骤一:pda培养基的制备
42.称取46g pda培养基粉末于100ml去离子水中并于高温灭菌锅中灭菌15min;
43.步骤二:病原菌接种
44.将pda平板不设置贴膜处理,将灭菌后的pda培养基于超净工作台倒入培养皿中,在培养皿中心接种立枯丝核菌病原菌,将样品在28℃培养箱中恒温培养7d,待菌丝长满平板后用备用;
45.步骤三:真菌dna的提取及pcr扩增
46.利用真菌dna提取试剂盒进行dna的提取,采用真菌rdna-its通用引物对菌株进行pcr扩增。
47.在一种优选的实施方式中,所述步骤二中接种的病原菌预先进行纯化,病原菌的纯化方法为:将病原真菌无污染的边缘菌丝挑至pda平板上培养,重复此步骤直至获得纯培养菌株,将培养的菌株在28℃培养箱中恒温培养7d。
48.在一种优选的实施方式中,所述步骤三中dna提取试剂盒为biomiga fungal gdna kit试剂盒,所述步骤三中真菌rdna-its通用引物为its1:5
’‑
tccgtaggtgaacctgcgg-3’,its4:5
’‑
tcctccgcttattgatatgg-3’。
49.在一种优选的实施方式中,所述步骤三中对对菌株进行pcr扩增时,pcr扩增程序
为94℃预变性3min,94℃变性30s,55℃退火45s,72℃延伸1min,共35个循环,反应总体系为25μl,包括2
×
taq pcr mix12.5μl、dna模板1μl、上下游引物各1μl,ddh2o 9.5μl。
50.实施例7:
51.与实施例6不同的是,步骤二中接种的病原菌为尖孢镰刀菌。
52.实施例8:
53.与实施例6不同的是,步骤二中接种的病原菌为黑孢菌。
54.实施例9:
55.与实施例6不同的是,步骤二中接种的病原菌为绿色木酶。
56.实施例10:
57.与实施例6不同的是,步骤二中接种的病原菌为燕麦镰刀菌。
58.dna浓度定性和定量分析:
59.(一)利用琼脂糖凝胶电泳对pcr产物进行定性分析
60.具体步骤为:(1)琼脂糖凝胶的制备:将原始tae溶液稀释50倍,取10mltae稀释溶液用去离子水定容至500ml,然后称取琼脂糖0.5g加入到50ml稀释后tae溶液中,于微波炉中加热90s至完全溶解,冷却降温后加入2ul的核酸染料充分混匀;
61.(2)灌胶:将冷却至60℃的琼脂糖溶液缓慢地注入一个带有“梳子”的胶床中(避免气泡产生),若有气泡产生用塑料移液管移去,待胶凝固后轻轻地移去“梳子”;
62.(3)上样:在疏孔中加入5ul待测的dna样品,5ulmarker作为对照。
63.(4)跑胶:将上好样的胶块放入电泳仪中,正确连接电泳槽和电源(加样孔一侧靠近阴极(黑末端)),设定好电压和时间(电压130mv,时间25min)。
64.(5)观察:电泳结束后取出胶板放到凝胶成像系统上进行观察,并拍照记录。
65.由图1(图中有膜和无膜处理的各条带从左到右分别为立枯丝核菌、尖孢镰刀菌、黑孢菌、绿色木酶、燕麦镰刀菌dna的凝胶电泳结果)凝胶电泳图结果显示有膜和无膜处理各样品均得到较为清晰的目的条带,且各条带单一,无明显拖尾现象,与2000的marker标准条带相比表明各样品核酸长度大约在600bp-700bp左右,且有膜处理的dna样品目的条带较无膜处理更为明显。
66.(二)利用微量分光光度计对dna浓度进行定量分析
67.利用微量分光光度计对dna的浓度进行测定,取真菌提取试剂盒中的ellution buffer 1ul作为对照,取待测真菌dna样品1ul,每个真菌dna设置4个重复,记录各样品及分别在波长260nm、280nm、230nm下的od值。
68.dna在波长为260nm处,有最大吸收峰,因此可用在260nm处的吸光度测定dna的浓度。此外,a260/a280,a260/a230的比值的大小以用来判断所提取dna的纯度,一般双链dna纯品的a260/a280应大于1.8,如果比值低于1.8或者2.0,表示存在蛋白质或酚类物质的影响,较纯净的a260/a230的比值一般在2.5左右,比值较低可能存在碳水化合物等其他污染物,由微量分光光度计测量结果表明贴了生物膜的真菌dna浓度显著高于不贴膜的真菌dna浓度,其中基于生物膜培养的燕麦镰刀菌提取的dna浓度最高,在260nm下的平均吸光度高达491.95ng/ul,其次为贴膜的黑孢菌、尖孢镰刀菌、立枯丝核菌,平均a260值分别为414.20、326.72和296.08ng/ul,而不贴生物膜培养的各真菌dna浓度均较低,其中最高的为尖孢镰刀菌的178.75ng/ul,立枯丝核菌dna浓度仅为28.65ng/ul,结果如表一所示,由此可
表明基于生物膜培养条件下可大大提高dna的提取效率。贴膜处理和无膜处理a260/a280的比值均大于1.8,各数值相差不大,没有显著性差异,表明各样品dna较为纯净。各样品a260/a230的比值均在2-2.6之间,各数值没有显著性差异,表明利用试剂盒法提取的样品dna均较纯。
[0069][0070][0071]
表一
[0072]
图中数据为平均数
±
标准差(每个实施例测试十组),分别比较有膜处理和无膜处理a260/a280和a260/a230的差异性,不同小写字母表示各处理间差异显著(p《0.05)。
[0073]
最后应说明的是:以上所述仅为本发明的优选实施例而已,并不用于限制本发明,尽管参照前述实施例对本发明进行了详细的说明,对于本领域的技术人员来说,其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1