索吡溴铵的制备方法与流程
本发明属于药物合成,具体涉及一种索吡溴铵的制备方法。
背景技术:
1、索吡溴铵,化学名为:(2r,3'r)-3'-(2-环戊基-2-羟基-2-苯基乙酰基)-1'-(乙氧基羰基甲基)-1'-甲基吡咯烷鎓溴化物。由下式a表示:
2、
3、在索吡溴铵的2-和3'-位置中的每一处的立体化学被鉴定为r-构型,但在1'-位置处季氮的立体化学未被鉴定。
4、更具体地,索吡溴铵是由下式(a-1)表示的(2r,3'r,1'r)-3'-(2-环戊基-2-羟基-2-苯基乙酰基)-1'-(乙氧基羰基甲基)-1'-甲基吡咯烷鎓溴化物:
5、
6、和由下式(a-2)表示的(2r,3'r,1's)-3'-(2-环戊基-2-羟基-2-苯基乙酰基)-1'-(乙氧基羰基甲基)-1'-甲基吡咯烷鎓溴化物,本发明的说明书其他地方的1'r-非对映异构体也被称为“化合物(a-1)”:
7、
8、组成的混合物,本发明的说明书其他地方的1's-非对映异构体被称为“化合物(a-2)”。
9、索吡溴铵是已知的可用于多汗症的治疗型处理的抗胆碱能剂,其被归类为可抑制神经递质乙酰胆碱作用的抗胆碱能药物。乙酰胆碱被认为通过与汗腺的毒蕈碱受体结合而引起出汗。索吡溴铵通过与内分泌汗腺中引起多汗症的毒蕈碱受体结合而抑制乙酰胆碱的结合,从而减少汗腺分泌。
10、在期刊pharmazie(2006),61(2),90-96(preparation and biological effectsof pure stereoisomeric novel soft anticholinergics),专利wo2007/05897公开了化合物(a)、(a-1)和(a-2)的制备方法。虽然其描述的方法可以分离纯化a-1和a-2,但纯度较低而且收率较低,而且没有对性质和晶型进行深入研究。专利wo2018026869a1公开甘草聚合物的制备方法和使用方法,虽然以上现有技术均公开了在乙腈中进行((2r,3'r)-3'-(2-环戊基-2-羟基-2-苯基乙酰氧基)-1'-甲基吡咯烷与溴乙酸甲酯的n-烷基化反应、然后将得到的粗制化合物(a)的二氯甲烷溶液加入乙醚以得到沉淀物的方法,但是其纯度较低,副产物较多,晶型不稳定,并且制备方法包括重复三次获得沉淀物的步骤,这样的合成方法难以应用于工业上,并且其中使用的二氯甲烷和乙醚不是工业上优选的溶剂。
11、依现有技术,控制n-1'-位置的立体选择性较为困难,所以得到的索吡溴铵为化合物(a),即一对非对映异构体(差向异构体)。而且分离化合物(a-1)和(a-2)的方法主要为普通柱层析法,实现工业化生产困难,且产率极低。专利cn114008023a公开了一种索吡溴铵的晶型及其制备方法,通过优化重结晶工艺,将化合物(a-1)和(a-2)的比例控制在1:2-1:3,获得物理化学稳定的晶型,作为药物的药用物质具有优异的性能,但难以实现工业化生产,并且该方法合成步骤繁琐,尤其是拆分dl-环戊基扁桃酸时,使用两种拆分剂会使收率较低;同时,在精制步骤中用到乙腈这一毒性较大的溶剂为精制溶剂,容易造成毒性溶剂残留,并且对环境不友好,不符合绿色化学的要求。
12、因此,寻找一种简单高效、环境友好的合成方法以实现索吡溴铵的工业化合成方法是非常有必要的。
技术实现思路
1、本发明要解决的技术问题是克服现有技术的不足,提供一种索吡溴铵的制备方法,步骤简单,避免了毒性溶剂的使用,降低环境污染的同时提高了收率,适合于工业化生产,具有较高的经济效益和现实意义。
2、本发明所述的索吡溴铵的制备方法,包括以下步骤:
3、(1)在(r/s)-α-环戊基扁桃酸中加入溶剂a加热溶解,继续加入l-酪氨酸甲酯,加热回流1h~3h,过滤浓缩后得到的滤液酸化至ph=1~2,冷却至室温,萃取后的有机相进行后处理,得到粗制的(r)-α-环戊基扁桃酸,继续将重复上述步骤,直至光学纯度(ee)不小于99.5%,得到(r)-α-环戊基扁桃酸,即为化合物i;其中优选的加入的l-酪氨酸甲酯分批加入,此步骤中的过滤浓缩后的滤液因状态为黄色油状物,需要对其进行进一步的溶解后再加热,均为实现其更高纯度的操作进行;
4、(2)在惰性气体保护下,将化合物i溶于溶剂b,在0℃~5℃下加入1,1'-羰基二咪唑反应,得到混合液a;将(r)-1-甲基-3-吡咯烷醇溶于溶剂b,在40℃~45℃加入叔丁醇钠反应,得到反应液b;将混合液a滴入混合液b中反应,至tlc检测反应反应物点消失,冷却至室温,经后处理,得到(r)-α-环戊基扁桃酸-(r)-1-甲基-3-吡咯烷醇配合物,记为化合物ⅱ;
5、(3)将化合物ⅱ溶于溶剂c,加入溴乙酸乙酯,在50℃~55℃下反应1.5h~2h,冷却至室温,得到的固体洗涤干燥,得到粗品化合物ⅲ;
6、(4)对粗品化合物ⅲ进行精制,得到索吡溴铵;
7、(5)利用柱层析对索吡溴铵分离纯化,得到高纯度a-1与a-2。
8、步骤(1)的后处理为采用无水硫酸钠干燥,过滤,滤液减压浓缩;溶剂a为乙腈、丙腈、丁腈、异丁腈中的一种或多种,优选为乙腈。调节ph采用硫酸、盐酸或混合酸,优选盐酸。
9、步骤(2)的后处理为加水稀释,调节ph至1~2,分液后水层调节ph至9~10,然后用乙酸乙酷萃取,减压浓缩;溶剂b为苯、甲苯中的一种或两种混合,优选甲苯。
10、步骤(3)的溶剂c为乙酸乙酯或甲基叔丁基醚。
11、步骤(1)的(r/s)-α-环戊基扁桃酸的摩尔量为l-酪氨酸甲酯的质量比为1:1.07~1:1.08。
12、步骤(2)的化合物i、1,1'-羰基二咪唑、(r)-1-甲基-3-吡咯烷醇、叔丁醇钠这四种物质的质量比为(13~14):(11~12):(18~19):1。
13、步骤(2)中,所述惰性保护气体为氩气、氮气,优选氮气。
14、骤(3)的化合物ⅱ与溴乙酸乙酯的质量比为1.7~1.8:1。
15、步骤(4)的精制步骤为:将粗品化合物ⅲ溶解在精制溶剂中,得到悬浊液;升温至50℃~55℃保温30min~31min,按照0.18ml/min~1.9ml/min的滴加速度,滴加甲基叔丁基醚50min~55min;降温至40℃~41℃,按照0.21ml/min~2.22ml/min的滴加速度,滴加甲基叔丁基醚44min~45min;降温至20℃~25℃,按照0.13ml/min~1.6ml/min的滴加速度,滴加甲基叔丁基醚35min~40min;降温至10℃~11℃,按照0.14ml/min~1.4ml/min的滴加速度,滴加甲基叔丁基醚35min~36min;降温至0℃~5℃,按照0.08ml/min~0.9ml/min的滴加速度,滴加甲基叔丁基醚30min~35min;搅拌,洗涤,干燥,得到索吡溴铵。
16、对于精制中的精制溶剂体积没有特别限制,只要可以溶解化合物ⅲ即可,但是使用的精制溶剂体积(ml)可以为化合物ⅲ质量(g)的2-15倍,优选4-12倍,更优选5-10倍。
17、精制溶剂为乙腈、甲基叔丁基醚、乙酸乙酯、乙醚、乙醇、氯仿、二氯甲烷、异丙醇中的一种或多种,优选乙醇和甲基叔丁基醚1:9,更优选乙酸乙酯和甲基叔丁基醚2:3。
18、步骤(5)柱层析所采用的硅胶为100目~200目、200目~300目、300目~400目;所采用的洗脱剂为甲醇、乙醇、二氯甲烷、氯仿、甲基叔丁基醚中的一种或多种,优选为甲醇和二氯甲烷混合,其比例为5:95~2:9,进一步优选为1:9~1:4。
19、具体的,所述的索吡溴铵的制备方法,包括以下步骤:
20、(1)在2.5l的反应瓶中加入(r/s)-α-环戊基扁桃酸,然后用乙腈加热溶解,继续加入l-酪氨酸甲酯(cdi),搅拌加热回流1h~3h,逐渐冷却至室温,滤除沉淀物,洗涤液与滤液合并后减压浓缩,得到的滤液为黄色油状物,将油状物用蒸馏水溶解,加热至60℃并搅拌0.5h,趁热过滤,待滤液温度降为40℃时,采用盐酸酸化至ph=1~2,冷却至室温,然后用乙酸乙酯萃取,萃取后的有机相进行用无水硫酸钠干燥,过滤,滤液减压浓缩,得到淡黄色固体的粗制的(r)-α-环戊基扁桃酸,检测其收率和ee值;
21、将得到的淡黄色固体溶于乙腈中,再次与l-酪氨酸甲酯反应2h,恢复至室温后处理方式如上,得淡黄色固体的粗制的(r)-α-环戊基扁桃酸。如此反复多次拆分,直至ee值不小于99.5%,得到(r)-α-环戊基扁桃酸,即为化合物i。
22、反应式如下:
23、
24、(2)在氮气气体保护下,将化合物i加入到250ml的反应瓶中,加入甲苯溶解,在0℃~5℃下加入1,1'-羰基二咪唑反应,得到混合液a;氮气保护下,在另一个250ml的反应瓶中加入(r)-1-甲基-3-吡咯烷醇,并用40ml甲苯溶解,溶解完全后加入叔丁醇钠,在40℃~45℃搅拌20min,得到反应液b;将混合液a滴入混合液b中反应,搅拌反应至tlc检测反应完成,自然冷却至室温,加水入蒸馏水搅拌,调节ph至1~2,分液后,水层用碳酸钾水溶液调ph至9~10,然后用乙酸乙酷萃取,减压浓缩,得无色油状物,即(r)-α-环戊基扁桃酸-(r)-1-甲基-3-吡咯烷醇配合物,记为化合物ⅱ。
25、反应式如下:
26、
27、(3)室温下,将化合物ⅱ溶于乙酸乙酯,先加小部分溴乙酸乙酯,出现白色沉淀后再加入剩余溴乙酸乙酯,在50℃~55℃下反应1.5h~2h,冷却至室温,将固体通过过滤进行收集,得到的固体用乙酸乙酯洗涤,干燥,得到白色固体,即粗品化合物ⅲ。其中的溴乙酸乙酯的分步加入操作为现有技术不再叙述。
28、反应式如下:
29、
30、(4)对粗品化合物ⅲ进行精制,在室温下,将粗品化合物ⅲ溶解在乙酸乙酯中,得到悬浊液;升温至50~55℃保温30~31min,按照0.18-1.9ml/min的滴加速度,滴加甲基叔丁基醚50~55min;降温至40~41℃,按照0.21~2.22ml/min的滴加速度,滴加甲基叔丁基醚44~45min;降温至20-25℃,按照0.13-1.6ml/min的滴加速度,滴加甲基叔丁基醚35-40min;降温至10~11℃,按照0.14-1.4ml/min的滴加速度,滴加甲基叔丁基醚35~36min;降温至0~5℃,按照0.08-0.9ml/min的滴加速度,滴加甲基叔丁基醚30-35min;搅拌,洗涤,干燥,得白色固体,得到索吡溴铵,即化合物a-1和a-2的混合物。
31、(5)利用柱层析对索吡溴铵分离纯化,选用300-400目硅胶,洗脱剂选用甲醇和二氯甲烷的混合溶剂,得到高纯度a-1与a-2。
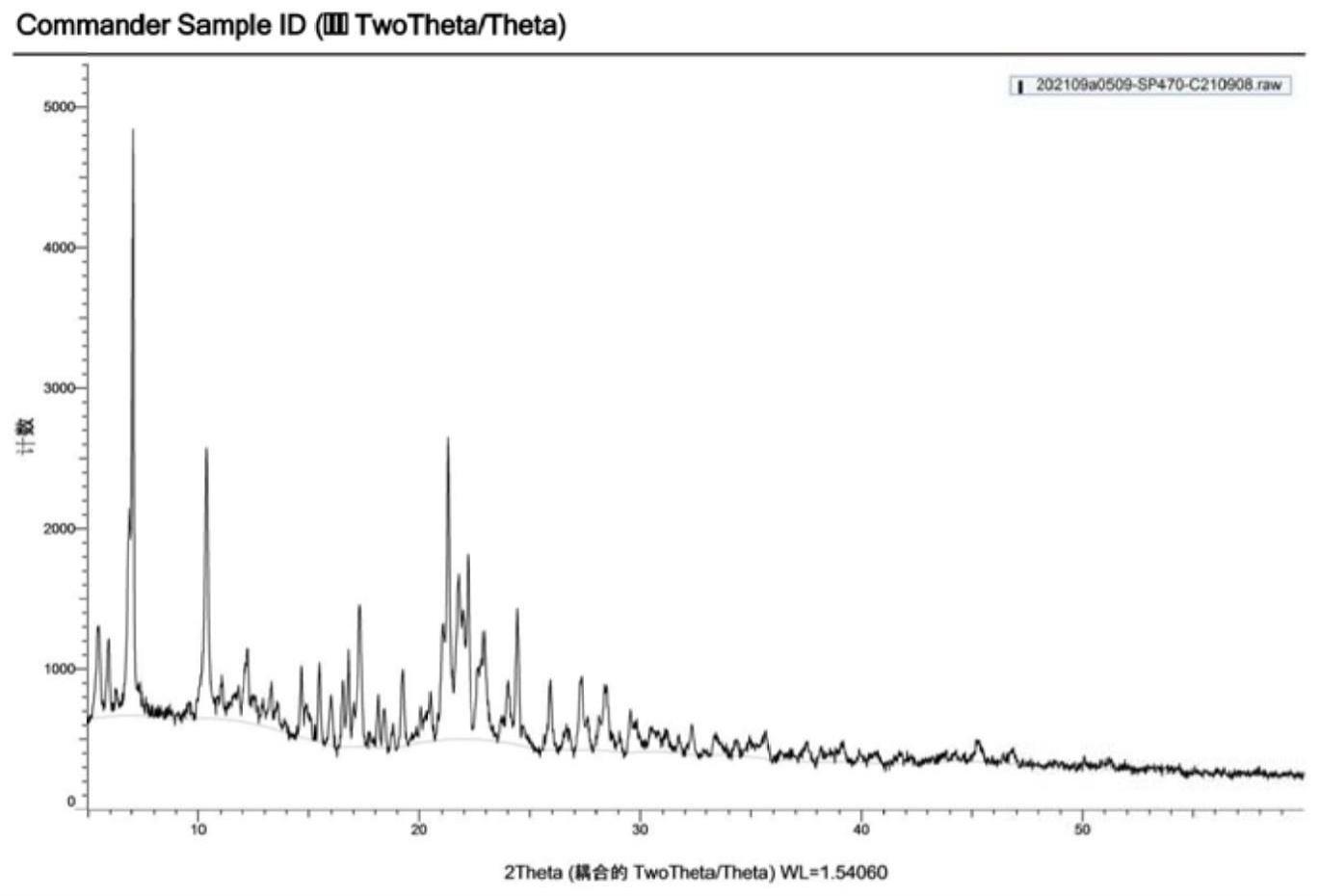
32、本发明的制备索吡溴铵的方法中,包括粗品化合物ⅲ的精制步骤,通过步骤(1)和步骤(2)所得的索吡溴铵为比例不固定的a-1和a-2的混合物,该混合物不具有适合于药物的药用物质的特性,通过精制步骤(3)后,可得到具有含有1:2比例的化合物(a-1与a-2)的共晶结构,而已有文献报道该比例的共晶结构作为药物的药用物质具有极其优异的性能。
33、与现有技术相比,本发明具有的有益效果是:
34、(1)本发明的索吡溴铵的制备方法,采取用l-酪氨酸甲酯多次拆分(r/s)-α-环戊基扁桃酸,得到(r)-α-环戊基扁桃酸,避免了更换拆分剂造成操作繁琐收率降低的弊端,相比使用两种拆分剂,收率明显提高,而且纯度较高,在工业化生产中简化步骤和操作的同时也能降低成本,提高收益。
35、(2)本发明的索吡溴铵的制备方法,采用特殊的精制条件,不但避免了具有较大毒性的溶剂乙腈的使用,也得到了对于药物的药用物质而言具有优异特性的晶型,同时还提高了收率和纯度。
36、(3)本发明的索吡溴铵的制备方法,在柱层析法中采用特定的洗脱剂以及其硅胶的选择,分离得到了高纯度的a-1和a-2,并对其进行了核磁表征,将收率提高到30%~40%。
- 还没有人留言评论。精彩留言会获得点赞!