一种阿昔替尼杂质M的合成方法与流程
一种阿昔替尼杂质m的合成方法
技术领域
1.本发明属于药物合成应用技术领域,具体涉及一种阿昔替尼杂质m的合成方法。
背景技术:
2.阿昔替尼(axitinib),化学名称n-甲基-2-[3-((e)-2-吡啶-2-基-乙烯基)-1h-吲哚-6-基磺酰]-苯甲酰胺,由美国辉瑞公司研发,于2012年1月在美国获得fda的批准。阿昔替尼是新的口服酪氨酸激酶抑制剂(tki),能有效并选择性抑制血管内皮生长因子受体vegfr-l、vegfr-2和vegfr-3,抑制血管和淋巴管的新生,抑制肿瘤的生长和转移,发挥抗肿瘤活性。该药通过阻断肿瘤生长过程中的蛋白激酶起到抑制肿块生长和癌症进展的作用。阿昔替尼,其分子式为c
22h18
n4os,分子量为386.47,cas号为319460-85-0,结构如下所示:
[0003][0004]
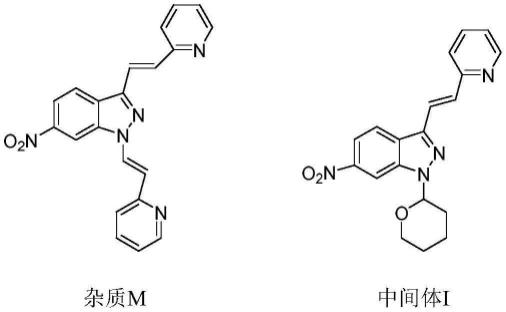
6-硝基-1,3-双((e)-2-(吡啶-2-基)乙烯基)-1h-吲唑(杂质m)是阿昔替尼中间体(e)-6-硝基-3-(2-(吡啶-2-基)乙烯基)-1-(四氢-2h-吡喃-2-基)-1h-吲唑(中间体i)制备过程中产生的杂质。药物纯度是由药物有效活性成分的含量决定的,而药物中存在的杂质直接影响到药物的疗效并可能导致毒副作用的产生。药物杂质是生产、储存过程中引进或产生的药物有效成分以外的其他化学物质,杂质的存在不仅影响药物的纯度,还会带来非治疗活性的毒副作用,必须加以控制。药物的质量标准对药物有效成分的纯度和杂质的限度都有较为严格的规定,一般而言,超过0.1%的药物杂质应通过选择性方法来鉴定并定量,对于药物研发人员来说,开发高效的杂质合成路线定向合成工艺中所产生的杂质,以便获得杂质对照品,保证每批原料药质量检测工作的开展(如,杂质hplc定位、杂质含量测定等)是十分重要的工作。随着国家对药品一致性研究工作的推进,确定阿昔替尼杂质的制备方法,提供合格的对照品,能够对阿昔替尼的质量控制起到积极的作用。
[0005][0006]
药品杂质能否被全面准确地控制,直接关系到药品的质量可控与安全有效。而目前杂质m的合成制备研究尚未见报道,因此,探究一种阿昔替尼杂质m的合成方法对杂质标准品的制备以及阿昔替尼产品的质量控制具有重要意义。
技术实现要素:
[0007]
本发明提供了一种阿昔替尼杂质m的合成方法,得到的对应杂质可为阿昔替尼的质量控制提供合格的对照品。
[0008]
本发明的具体技术方案如下:
[0009]
一种阿昔替尼杂质m的合成方法,其特征在于,包括以下步骤:以sm-1为起始原料,与2-乙炔基吡啶反应,制得杂质m,反应式如下:
[0010][0011]
优选地,所述阿昔替尼杂质m的合成方法,具体包括以下步骤:惰性气体保护下,将sm-1、2-乙炔基吡啶和催化剂加入反应溶剂中,控温反应至反应结束,制得杂质m。
[0012]
优选地,所述催化剂选自硝酸银、碘化亚铜、溴化亚铜中的一种,优选硝酸银。
[0013]
优选地,所述反应溶剂选自dmso、dmf中的一种,优选dmso。
[0014]
优选地,所述控温温度为120~150℃,优选130~135℃。
[0015]
优选地,所述2-乙炔基吡啶与sm-1的投料摩尔比为1:1.5~4,优选1:2。
[0016]
优选地,所述2-乙炔基吡啶与催化剂的投料摩尔比为1:0.025~0.1,优选1:0.05。
[0017]
所述惰性气体为氮气、氩气中的一种。
[0018]
在一优选方案中,所述合成步骤还包括对制得的杂质m进行分离纯化,具体为:将反应液降至室温,加入饱和nahco3溶液,搅拌,加入适量有机溶剂和水,分液取有机相,浓缩,所得浓缩物用制备hplc纯化,收集制备接出液,浓缩、冻干得固体杂质m。优选地,所述有机溶剂包括但不限于乙酸乙酯、二氯甲烷中的一种。
[0019]
所述制备hplc纯化,具体包括以下步骤:将浓缩物溶于乙醇中作为样品储备液;取储备液,用流动相稀释,超声溶解,过滤,用液相色谱梯度洗脱分离,收集制备接出液。
[0020]
进一步地,所述制备hplc纯化,具体实施方式包括:(1)配制制备液:将浓缩物溶于乙醇中作为样品储备液,取储备液,用流动相稀释,超声溶解,过滤,配制制备液;(2)制备条件:设备为汉邦dac-50动态轴向压缩系统,以c18为填料,波长为250~280nm,柱温为常温,流速为50~70ml/min,流动相梯度洗脱,接出收集洗脱液累积。
[0021]
所述样品储备液浓度没有严格限制,但乙醇的量要至少能够溶解浓缩物。
[0022]
所述流动相稀释后的浓度同样没有严格限制,但稀释后的溶液要能够完全溶解样品。
[0023]
优选地,所述流动相为乙腈和水,其中配制制备液时流动相中乙腈和水的体积比为1:1.05~1.3,优选1:1.15。
[0024]
优选地,所述洗脱梯度为:0~25min,60%~40%水;25~30min,30%~15%水;30~35min,40%~55%水。
[0025]
更优选地,所述洗脱梯度为:0~25min,50%~40%水;25~30min,25%~20%水;30~35min,40%~45%水。
[0026]
更进一步优选地,所述洗脱梯度具体为:
[0027]
t(min)水乙腈0~2545.055.025~3020.080.030~3545.055.0
[0028]
本发明的有益效果:本发明提供了一种阿昔替尼杂质m的合成方法,该方法以sm-1(e型)和2-乙炔基吡啶为原料,制备得到固体状态的阿昔替尼杂质m(e型),解决了杂质m固体纯品制备的问题;该方法简单,制得的杂质m收率高、纯度好、易于分离,将本发明所制得的杂质m纯品作为外标物,为阿昔替尼的质量研究提供基础。
附图说明
[0029]
图1:阿昔替尼杂质m一次制备接出液检测图。
[0030]
图2:阿昔替尼杂质m的红外图谱。
[0031]
图3:阿昔替尼杂质m的质谱图。
[0032]
图4:阿昔替尼杂质m的核磁氢谱。
[0033]
图5:阿昔替尼杂质m的noesy谱。
[0034]
图6:阿昔替尼杂质m的cosy谱。
[0035]
图7:阿昔替尼杂质m的光谱图。
具体实施方式
[0036]
下面通过实施例来进一步说明本发明,应该正确理解的是:本发明的实施例仅仅是用于说明本发明,而不是对本发明的限制,所以,在本发明的方法前提下对本发明的简单改进均属于本发明要求保护的范围。
[0037]
本发明制备方法所用起始原料sm-1(e型)和2-乙炔基吡啶可根据常规方法制备或购买使用,以下各实施例中,未详细描述的各种过程和方法是本领域中公知的常规方法。
[0038]
实施例1
[0039]
氮气保护下,将sm-1(0.500mmol)、2-乙炔基吡啶(0.250mmol)和agno3(12.5μmol)加入至dmso(10ml)中,控温130~135℃下搅拌反应至反应结束后,将反应液降至室温,加入饱和nahco3水溶液(1ml),搅拌,加入乙酸乙酯(30ml),用纯化水(30ml
×
3)冲洗有机相,分液取有机相,浓缩,所得浓缩物溶于乙醇中作为样品储备液,取储备液,加入乙腈和水(v
乙腈
:v
水
=1:1.15)稀释,超声溶解,过滤,将制备液用汉邦dac-50动态轴向压缩系统,进行分离纯化,洗脱梯度设置为:0-25分钟,45%水;25-30分钟,20%水;30-35分钟,45%水;收集25~28.5分钟的洗脱液累积,35℃旋蒸浓缩至产品析出,过滤,冻干得42.2mg固体杂质m,纯度98.19%。
[0040]
实施例2
[0041]
氮气保护下,将sm-1(1.0mmol)、2-乙炔基吡啶(0.250mmol)和agno3(12.5μmol)加入至dmso(10ml)中,控温120~125℃下搅拌反应至反应结束后,将反应液降至室温,加入饱和nahco3水溶液(1ml),搅拌,加入二氯甲烷(30ml),用纯化水(30ml
×
3)冲洗有机相,分液取有机相,浓缩,所得浓缩物溶于乙醇中作为样品储备液,取储备液,加入乙腈和水(v
乙腈
:v
水
=1:1.15)稀释,超声溶解,过滤,将制备液用汉邦dac-50动态轴向压缩系统,进行分离纯化,洗脱梯度设置为:0-25分钟,45%水;25-30分钟,20%水;30-35分钟,45%水;收集25~28.5分钟的洗脱液累积,35℃旋蒸浓缩至产品析出,过滤,冻干得39.8mg固体杂质m,纯度98.05%。
[0042]
实施例3
[0043]
氮气保护下,将sm-1(0.375mmol)、2-乙炔基吡啶(0.250mmol)和agno3(12.5μmol)加入至dmso(10ml)中,控温145~150℃下搅拌反应至反应结束后,将反应液降至室温,加入饱和nahco3水溶液(1ml),搅拌,加入乙酸乙酯(30ml),用纯化水(30ml
×
3)冲洗有机相,分液取有机相,浓缩,所得浓缩物溶于乙醇中作为样品储备液,取储备液,加入乙腈和水(v
乙腈
:v
水
=1:1.15)稀释,超声溶解,过滤,将制备液用汉邦dac-50动态轴向压缩系统,进行分离纯化,洗脱梯度设置为:0-25分钟,45%水;25-30分钟,20%水;30-35分钟,45%水;收集25~28.5分钟的洗脱液累积,35℃旋蒸浓缩至产品析出,过滤,冻干得35.2mg固体杂质m,纯度98.0%。
[0044]
实施例4
[0045]
氩气保护下,将sm-1(0.500mmol)、2-乙炔基吡啶(0.250mmol)和cui(25μmol)加入至dmf(10ml)中,控温130~135℃下搅拌反应至反应结束后,将反应液降至室温,加入饱和nahco3水溶液(1ml),搅拌,加入乙酸乙酯(30ml),用纯化水(30ml
×
3)冲洗有机相,分液取有机相,浓缩,所得浓缩物溶于乙醇中作为样品储备液,取储备液,加入乙腈和水(v
乙腈
:v
水
=1:1.15)稀释,超声溶解,过滤,将制备液用汉邦dac-50动态轴向压缩系统,进行分离纯化,洗脱梯度设置为:0-25分钟,45%水;25-30分钟,20%水;30-35分钟,45%水;收集25~28.5分钟的洗脱液累积,35℃旋蒸浓缩至产品析出,过滤,冻干得33.8mg固体杂质m,纯度98.08%。
[0046]
实施例5
[0047]
氩气保护下,将sm-1(0.500mmol)、2-乙炔基吡啶(0.250mmol)和cubr(6.25μmol)加入至dmf(10ml)中,控温130~135℃下搅拌反应至反应结束后,将反应液降至室温,加入饱和nahco3水溶液(1ml),搅拌,加入乙酸乙酯(30ml),用纯化水(30ml
×
3)冲洗有机相,分
液取有机相,浓缩,所得浓缩物溶于乙醇中作为样品储备液,取储备液,加入乙腈和水(v
乙腈
:v
水
=1:1.15)稀释,超声溶解,过滤,将制备液用汉邦dac-50动态轴向压缩系统,进行分离纯化,洗脱梯度设置为:0-25分钟,45%水;25-30分钟,20%水;30-35分钟,45%水;收集25~28.5分钟的洗脱液累积,35℃旋蒸浓缩至产品析出,过滤,冻干得29.1mg固体杂质m,纯度97.85%。
[0048]
实施例6
[0049]
氩气保护下,将sm-1(0.500mmol)、2-乙炔基吡啶(0.250mmol)和agno3(25μmol)加入至dmf(10ml)中,控温130~135℃下搅拌反应至反应结束后,将反应液降至室温,加入饱和nahco3水溶液(1ml),搅拌,加入乙酸乙酯(30ml),用纯化水(30ml
×
3)冲洗有机相,分液取有机相,浓缩,所得浓缩物溶于乙醇中作为样品储备液,取储备液,加入乙腈和水(v
乙腈
:v
水
=1:1.15)稀释,超声溶解,过滤,将制备液用汉邦dac-50动态轴向压缩系统,进行分离纯化,洗脱梯度设置为:0-25分钟,45%水;25-30分钟,20%水;30-35分钟,45%水;收集25~28.5分钟的洗脱液累积,35℃旋蒸浓缩至产品析出,过滤,冻干得40.0mg固体杂质m,纯度98.02%。
[0050]
实施例7
[0051]
氮气保护下,将sm-1(1.0mmol)、2-乙炔基吡啶(0.250mmol)和agno3(12.5μmol)加入至dmso(10ml)中,控温120~125℃下搅拌反应至反应结束后,将反应液降至室温,加入饱和nahco3水溶液(1ml),搅拌,加入二氯甲烷(30ml),用纯化水(30ml
×
3)冲洗有机相,分液取有机相,浓缩,所得浓缩物溶于乙醇中作为样品储备液,取储备液,加入乙腈和水(v
乙腈
:v
水
=1:1.15)稀释,超声溶解,过滤,将制备液用汉邦dac-50动态轴向压缩系统,进行分离纯化,洗脱梯度设置为:0-25分钟,45%水;25-30分钟,20%水;30-35分钟,45%水;收集25~28.5分钟的洗脱液累积,35℃旋蒸浓缩至产品析出,过滤,烘干得38.5mg油状杂质m,纯度97.90%。
[0052]
实施例8
[0053]
氩气保护下,将sm-1(0.500mmol)、2-乙炔基吡啶(0.250mmol)和agno3(25μmol)加入至dmf(10ml)中,控温130~135℃下搅拌反应至反应结束后,将反应液降至室温,加入饱和nahco3水溶液(1ml),搅拌,加入乙酸乙酯(30ml),用纯化水(30ml
×
3)冲洗有机相,分液取有机相,浓缩,所得浓缩物溶于乙醇中作为样品储备液,取储备液,加入乙腈和水(v
乙腈
:v
水
=1:1.3)稀释,超声溶解,过滤,将制备液用汉邦dac-50动态轴向压缩系统,进行分离纯化,洗脱梯度设置为:0-25分钟,40%水;25-30分钟,30%水;30-35分钟,55%水;收集25~28.5分钟的洗脱液累积,35℃旋蒸浓缩至产品析出,过滤,冻干得35.6mg固体杂质m,纯度97.94%。
[0054]
实施例9
[0055]
氩气保护下,将sm-1(0.500mmol)、2-乙炔基吡啶(0.250mmol)和agno3(25μmol)加入至dmf(10ml)中,控温130~135℃下搅拌反应至反应结束后,将反应液降至室温,加入饱和nahco3水溶液(1ml),搅拌,加入乙酸乙酯(30ml),用纯化水(30ml
×
3)冲洗有机相,分液取有机相,浓缩,所得浓缩物溶于乙醇中作为样品储备液,取储备液,加入乙腈和水(v
乙腈
:v
水
=1:1.05)稀释,超声溶解,过滤,将制备液用汉邦dac-50动态轴向压缩系统,进行分离纯化,洗脱梯度设置为:0-25分钟,50%水;25-30分钟,25%水;30-35分钟,40%水;收集25~
28.5分钟的洗脱液累积,35℃旋蒸浓缩至产品析出,过滤,冻干得32.9mg固体杂质m,纯度97.88%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1