液晶取向剂、液晶取向膜、液晶元件、液晶取向膜的制造方法以及倍半硅氧烷与流程
本发明涉及一种液晶取向剂、液晶取向膜、液晶元件、液晶取向膜的制造方法以及化合物,尤其涉及一种液晶取向剂、液晶取向膜、液晶元件、液晶取向膜的制造方法以及倍半硅氧烷。
背景技术:
液晶元件包括控制液晶分子的取向的液晶取向膜。构成液晶取向膜的材料已知聚酰亚胺、聚酰胺、聚酯、聚硅氧烷等,包含这些材料中的聚酰亚胺的液晶取向膜由于耐热性、机械强度以及与液晶分子的亲和性优异等,故而被使用已久(例如参照专利文献1)。另外,近年来,由于耐光性及耐热性良好等的原因,使用含有使硅烷化合物进行反应而获得的聚硅氧烷的液晶取向剂的情况也增加(例如参照专利文献2及专利文献3)。专利文献2中公开了:使用将烷氧基硅烷化合物在草酸的存在下进行缩聚而获得的聚硅氧烷来作为液晶取向剂的聚合体成分。另外,专利文献3中公开了如下的液晶取向剂,其含有经过在碱金属化合物或有机碱的存在下将硅烷化合物进行水解或水解·缩合的步骤而合成的聚硅氧烷、其水解物或者水解物的缩合物。
[现有技术文献]
[专利文献]
[专利文献1]日本专利特开2010-97188号公报
[专利文献2]日本专利第5593611号公报
[专利文献3]国际公开第2009/025388号
技术实现要素:
[发明所要解决的问题]
依据所述专利文献2中记载的方法而获得无规型的聚硅氧烷,依据所述专利文献3中记载的方法而获得笼型的聚硅氧烷。然而,无规型或笼型的聚硅氧烷的极限分子量低,在于基板上形成涂膜的情况下会产生涂布不均,由此存在制品良率下降、或显示品质下降的顾虑。随着近年来的液晶面板的高精细化,对显示品质的要求越来越严,作为液晶元件,期望电气特性及液晶取向性的进一步改善。
本发明是鉴于所述情况而形成,目的之一在于提供一种对基板的涂布性良好、且可获得液晶取向性及电气特性良好的液晶元件的液晶取向剂。
[解决问题的技术手段]
依据本发明来提供以下的手段。
[1]一种液晶取向剂,其含有具有下述式(1)所表示的梯型结构的倍半硅氧烷[L],
[化1]
(式(1)中,E1及E2分别独立地为一价有机基,n为2以上的整数;不同的重复单元中的E1及E2可相同也可不同)。
[1-1]一种液晶取向剂,其含有满足下述的必要条件(A)~(C)的倍半硅氧烷[L],
(A)利用凝胶渗透色谱法(Gel Permeation Chromatography,GPC)来测定的重量平均分子量(Mw)为5000以上;
(B)利用GPC来测定的由重量平均分子量(Mw)与数量平均分子量(Mn)的比所表示的分子量分布(Mw/Mn)为3.8以下;
(C)由下述式(4)所表示的部分结构而来的29Si-NMR光谱的峰值的积分比为70%~99%;
[化21
(式(4)中,Ra为碳数1以上的一价有机基,“*”表示与硅原子的结合键)。
[2]一种液晶取向剂,其含有在满足下述的条件1、条件2及条件3中的两个以上的条件下将硅烷化合物聚合而获得的倍半硅氧烷[L],
条件1:包含具有一种或两种以上的如下特定基的硅烷化合物来作为所述硅烷化合物,所述特定基选自由-NHR5(其中,R5为氢原子或碳数1~6的烷基;以下相同)、吡啶基、咪唑基、氰基、亚氨基、羟基、巯基、羧基、磷酰基、磺基及酸酐基所组成的群组中;
条件2:在所述条件1的硅烷化合物具有酸性基作为所述特定基的情况下,在碱的存在下将所述硅烷化合物水解缩合,在具有碱性基作为所述特定基的情况下,在酸的存在下将所述硅烷化合物水解缩合;
条件3:在包含10质量%以上的水的溶媒中将所述硅烷化合物水解缩合。
[3]一种液晶取向膜,其是使用所述[1]、[1-1]或[2]的液晶取向剂而形成。
[4]一种液晶元件,其包括所述[3]的液晶取向膜。
[5]一种液晶取向膜的制造方法,其包括:将所述[1]、[1-1]或[2]的液晶取向剂涂布于基板上而形成涂膜的步骤;以及对涂布有该液晶取向剂的基板面进行光照射的步骤。
[6]一种倍半硅氧烷,其具有所述式(1)所表示的梯型结构;其中,倍半硅氧烷所具有的多个E1及E2中的一个以上为具有选自由以下基团所组成的群组中的至少一种的基团:环氧基、氧杂环丁基、酸酐基、(甲基)丙烯酰基、苯乙烯基、乙炔基、巯基、异氰酸酯基、醇性羟基、氰基、-COOR2、-CON(R1R2)、-PO(R1)2、-SO3R2、-SO2N(R1R2)(其中,R1分别独立地为氢原子或一价烃基,R2为一价烃基)、光取向性基及交联性基。
[发明的效果]
依据所述包含倍半硅氧烷[L]的液晶取向剂,可获得对基板的涂布性良好的液晶取向剂。另外,可获得液晶取向性及电气特性良好的液晶元件。
附图说明
图1是实施例中合成的聚合体的29Si-NMR光谱。
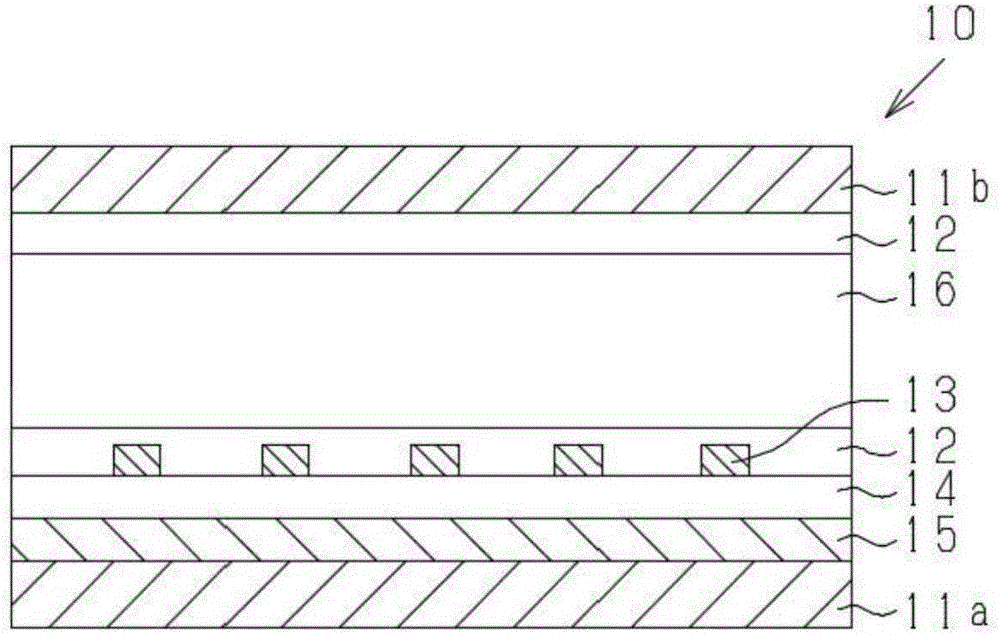
图2是边缘场切换(Fringe Field Switching,FFS)型液晶单元的概略构成图。
图3(a)及图3(b)是用于通过光取向处理来制造液晶显示元件的顶电极的平面示意图。图3(a)为顶电极的俯视图,图3(b)为顶电极的部分放大图。
图4是表示四系统的驱动电极的图。
附图标记:
10:液晶显示元件
11a、11b:基板
12:液晶取向膜
13:顶电极
14:绝缘层
15:底电极
16:液晶层
A、B、C、D:电极
C1:虚线包围的部分
d1:电极的线宽
d2:电极间的距离
具体实施方式
以下,对本发明的液晶取向剂中所含的各成分、以及视需要而任意调配的其他成分进行说明。
<倍半硅氧烷[L]>
本发明的液晶取向剂含有具有所述式(1)所表示的梯型结构的倍半硅氧烷[L]。
所述式(1)中,E1及E2的一价有机基优选为包含至少一个碳原子的基团,也可在结构中包含杂原子。作为一价有机基的具体例,例如可列举:碳数1~20的一价烃基、在该烃基的碳-碳键间包括含杂原子的基的基团、该烃基与含杂原子的基键结而成的基团、将这些基团的至少一个氢原子由取代基取代而成的基团等。
此处,本说明书中,所谓“有机基”是可构成有机化合物的一部分的基团,也可在结构中包含杂原子。该有机基为包含官能基(例如氨基等)的概念。所谓“含杂原子的基”只要是具有杂原子的基团即可,例如可列举:-O-、-CO-、-COO-、-CONRa-(Ra为氢原子或碳数1~6的烷基,以下相同)、-NRa-、三价氮原子、-NRaCONRa-、-OCONRa-、-S-、-COS-、-OCOO-、-SO2-、磷酰基等。取代基例如可列举:卤素原子、硝基、氰基、羟基、氨基、吡啶基、咪唑基、巯基、羧基、磷酸基、磺基、(甲基)丙烯酰基等。此外,“(甲基)丙烯酰”是指包含“丙烯酰”及“甲基丙烯酰”。
所谓“烃基”是包含链状烃基、脂环式烃基及芳香族烃基的含义。所谓“链状烃基”是指在主链上不包含环状结构,而仅由链状结构所构成的直链状烃基及分支状烃基。其中,可为饱和,也可为不饱和。所谓“脂环式烃基”是指仅包含脂环式烃的结构作为环结构,而不包含芳香环结构的烃基。其中,不需要仅由脂环式烃的结构所构成,也包含在其一部分中具有链状结构的。所谓“芳香族烃基”是指包含芳香环结构作为环结构的烃基。其中,不需要仅由芳香环结构所构成,也可在其一部分中包含链状结构或脂环式烃的结构。
作为E1及E2,优选为倍半硅氧烷[L]所具有的多个E1及E2的至少一个具有选自由以下基团所组成的群组中的至少一种:环氧基、氧杂环丁基、酸酐基、(甲基)丙烯酰基、乙烯基(烯基、苯乙烯基中的乙烯基等)、乙炔基、巯基、-NHR5、异氰酸酯基、醇性羟基、酚性羟基、氰基、-COOR1、-CON(R1)2、-PO(R1)2、-SO3R1及-SO2N(R1)2(其中,R1分别独立地为氢原子或一价烃基)。R1优选为氢原子或碳数1~10的烷基,更优选为氢原子或碳数1~5的烷基。
倍半硅氧烷[L]所具有的多个E1及E2中的一个以上优选为具有选自由具有预倾角表现能力的基团、光取向性基及交联性基所组成的群组中的至少一种。通过设为在侧链上具有所述基团,可对所获得的有机膜赋予与各基团对应的功能。特别是具有所述式(1)所表示的梯型结构的倍半硅氧烷与例如笼型或无规型的相比,在能够任意地控制具有预倾角表现能力的基团或光取向性基、交联性基等功能性侧链的导入数,容易调节取向方位的控制性或液晶元件的可靠性的方面优选。
具有预倾角表现能力的基团是可不通过光照射,而对涂膜赋予预倾角特性的基团,具体而言,例如可列举:碳数3~20的烷基、碳数3~20的氟烷基、碳数3~20的烷氧基、碳数17~51的具有类固醇骨架的基团、具有两个以上的环直接或者经由连结基而连结的结构(以下也称为“多环结构”)的基团等。作为这些基团的具体例,碳数3~20的烷基例如可列举:正丙基、正丁基、正戊基、正己基、正庚基、正辛基、正壬基、正癸基、正十二烷基、正十三烷基、正十四烷基、正十五烷基、正十六烷基、正十七烷基、正十八烷基、正十九烷基、正二十烷基等;碳数3~20的氟烷基例如可列举:将所述例示的烷基的至少一个氢原子由氟原子取代而成的基团等;碳数3~20的烷氧基例如可列举:在所述例示的烷基上键结有氧原子的基团等;碳数17~51的具有类固醇骨架的基团例如可列举:具有胆甾烷基、胆甾烯基或者羊毛甾烷基的基团等;具有多环结构的基团例如可列举:具有4,4′-亚联苯基、4,4′-亚双环己基(bicyclohexylene)以及下述式(6-1)~式(6-4)
[化3]
(式中,“*”表示结合键)
各自所表示的基团的基团等。倍半硅氧烷[L]所具有的具有预倾角表现能力的基团可仅为一种,也可为两种以上。
光取向性基只要是通过光异构化或光二聚化等而显示出光取向性的基团(光反应性基)即可。作为光取向性基的具体例,例如可列举:将偶氮苯或其衍生物作为基本骨架的含偶氮苯的基团、将肉桂酸或其衍生物作为基本骨架的含肉桂酸结构的基团、将查耳酮或其衍生物作为基本骨架的含查耳酮的基团、将二苯甲酮或其衍生物作为基本骨架的含二苯甲酮的基团、将香豆素或其衍生物作为基本骨架的含香豆素的基团等。这些基团中优选为含肉桂酸结构的基团。
交联性基优选为可利用光或热而在同一或不同的分子间形成共价键的基团,例如可列举:将(甲基)丙烯酸或其衍生物作为基本骨架的含(甲基)丙烯酸基的基团、具有乙烯基的基团(烯基、苯乙烯基等)、乙炔基、环氧基、氧杂环丁基等。其中,优选为含(甲基)丙烯酸基的基团、苯乙烯基。
此外,倍半硅氧烷[L]可仅包含具有预倾角表现能力的基团、光取向性基及交联性基中的任一者,也可包含这些基团的两种以上。另外,也可在一个E1或一个E2中包含具有预倾角表现能力的基团、以及(甲基)丙烯酰基或乙烯基等交联性基。所述基团显示出预倾角表现能力,并且显示出交联性。所述式(1)中的n为2以上,优选为5以上,更优选为8以上。关于n的上限值,可根据所需的分子量来适宜选择。
(倍半硅氧烷[L]的合成)
倍半硅氧烷[L]的合成方法只要获得具有所述式(1)所表示的结构的倍半硅氧烷,则并无特别限定。优选为倍半硅氧烷[L]为水解性的硅烷化合物的水解缩合物。就所获得的倍半硅氧烷的结构规则性高的方面、或分子量及分子量分布的控制性高的方面而言,优选为利用:在满足下述的条件1、条件2及条件3中的两个以上的条件下将硅烷化合物进行聚合的方法(以下称为“方法1”)、或者将包含下述式(3)所表示的化合物的硅烷化合物进行聚合的方法(以下称为“方法2”)。
条件1:包含具有一种或两种以上的以下特定基的硅烷化合物来作为硅烷化合物,所述特定基选自由-NHR5、吡啶基、咪唑基、氰基、亚氨基、羟基、巯基、羧基、磷酰基、磺基及酸酐基所组成的群组中。
条件2:在所述条件1的硅烷化合物具有酸性基作为所述特定基的情况下,在碱的存在下将所述硅烷化合物水解缩合,在具有碱性基作为所述特定基的情况下,在酸的存在下将所述硅烷化合物水解缩合。
条件3:在包含10质量%以上的水的溶媒中将硅烷化合物水解缩合。
[化4]
J1-Ar1-Si(OR4)3 (3)
(式(3)中,Ar1为选自由下述式(ar-1)~式(ar-8)各自所表示的基团所组成的群组中的一种,J1为选自由下述式(j-1)~式(j-7)各自所表示的基团所组成的群组中的一种;R4为碳数1~18的一价烃基)
[化5]
(式(ar-1)~式(ar-8)中,“*”表示键结于硅原子上的结合键)
[化6]
(式(i-1)~式(j-7)中,R6为单键或碳数1~5的烷二基,R7为碳数1~5的烷基,X4为氢原子或碳数1~6的烷基;“*”表示与Ar1的结合键)
方法1中,聚合中使用的硅烷化合物优选为包含具有极性基的硅烷化合物,特别优选为包含具有一种或两种以上的如下特定基的硅烷化合物(以下也称为“特定硅烷化合物”),所述特定基选自-NHR5、吡啶基、咪唑基、氰基、亚氨基、羟基、巯基、羧基、磷酰基、磺基及酸酐基中。特定硅烷化合物的优选例可列举下述式(2)所表示的化合物。
[化7]
(式(2)中,R3为碳数2~18的有机基,所述有机基具有一种或两种以上的选自由-NHR5、吡啶基、咪唑基、氰基、亚氨基、羟基、巯基、羧基、磷酰基、磺基及酸酐基所组成的群组中的特定基;X1、X2及X3分别独立地为卤素原子、碳数1~18的烷氧基或碳数1~18的酰氧基)
所述式(2)的X1、X2及X3中的卤素原子例如可列举:氟原子、氯原子、溴原子、碘原子等。碳数1~18的烷氧基可列举所述E1及E2的说明中所例示的基团。碳数1~18的酰氧基例如可列举:乙酰氧基、丙酰氧基、苯甲酰氧基等。作为X1、X2及X3,这些基团中,优选为卤素原子、甲氧基或乙氧基。此外,X1、X2及X3可相互相同也可不同。
作为所述式(2)所表示的化合物,可列举选自由以下化合物所组成的群组中的至少一种化合物来作为优选的具体例:2-氰基乙基三乙氧基硅烷、3-氨基丙基三乙氧基硅烷、3-巯基丙基三乙氧基硅烷、1-甲基-3-(3-三甲氧基硅烷基丙基)-1H-咪唑-3-鎓氯化物、(3-苯甲酰氧基丙基)三甲氧基硅烷、三甲氧基硅烷基丙基乙酰胺、4-(2-(三甲氧基硅烷基)乙基)苯磺酸乙酯、N-(3-丙基三甲氧基硅烷)咪唑、N-(3-三甲氧基硅烷基丙基)天冬氨酸、3-(间二甲基氨基苯氧基)丙基三甲氧基硅烷、3-(三甲氧基硅烷基)丙酸甲酯、以及下述式(2-1)所表示的化合物。
[化8]
此外,在将特定硅烷化合物用于聚合的情况下,可将特定硅烷化合物添加于聚合体系中来进行聚合,或者也可通过体系内的反应来制备特定硅烷化合物而进行聚合。
倍半硅氧烷[L]的合成中使用的硅烷化合物可单独使用如上所述的特定硅烷化合物,但也可与特定硅烷化合物一起并用其他的硅烷化合物。其他的硅烷化合物若为水解性的硅烷化合物即可,作为具体例,例如可列举:四甲氧基硅烷、四乙氧基硅烷、三甲基乙氧基硅烷、甲基三甲氧基硅烷、二甲基二乙氧基硅烷、苯基三甲氧基硅烷、苯基三乙氧基硅烷、二甲基二甲氧基硅烷、十二烷基三乙氧基硅烷、十八烷基三乙氧基硅烷、十三氟辛基三甲氧基硅烷、4-(氯甲基)苯基三甲氧基硅烷等烷氧基硅烷化合物;
3-缩水甘油氧基丙基三甲氧基硅烷、3-缩水甘油氧基丙基三乙氧基硅烷、3-缩水甘油氧基丙基甲基二甲氧基硅烷、2-(3,4-环氧基环己基)乙基三甲氧基硅烷等含环氧基的硅烷化合物;
3-(甲基)丙烯酰氧基丙基三甲氧基硅烷、3-(甲基)丙烯酰氧基丙基三乙氧基硅烷、3-(甲基)丙烯酰氧基丙基甲基二甲氧基硅烷、3-(甲基)丙烯酰氧基丙基甲基二乙氧基硅烷、乙烯基三甲氧基硅烷、乙烯基三乙氧基硅烷、对苯乙烯基三甲氧基硅烷等含不饱和键的烷氧基硅烷化合物;
1,6-双(三甲氧基硅烷基)己烷、三-(3-三甲氧基硅烷基丙基)异氰脲酸酯、1,8-双(三甲氧基硅烷基)辛烷、双[3-(三甲氧基硅烷基)丙基]胺、三乙氧基硅烷基硫丙基三甲氧基硅烷等包含两个以上硅原子的硅烷化合物等。其他的硅烷化合物可单独使用这些化合物中的一种或者将两种以上组合使用。
合成倍半硅氧烷[L]时,相对于倍半硅氧烷[L]的合成中使用的硅烷化合物的合计,其他的硅烷化合物的使用比例优选为设为50摩尔%以下,更优选为设为40摩尔%以下。
方法1的反应是通过使如上所述的硅烷化合物的一种或两种以上,优选为在催化剂及溶媒的存在下进行水解·缩合而进行。关于反应中使用的催化剂,在使用具有氨基等碱性基的硅烷化合物作为特定硅烷化合物的情况下优选使用酸,特别优选为盐酸。另一方面,在使用具有羧基等酸性基的硅烷化合物作为特定硅烷化合物的情况下,优选使用碱作为催化剂,更优选为碱金属化合物,特别优选为氢氧化钠。催化剂的使用比例应根据催化剂的种类、温度等反应条件等来适宜设定,例如相对于硅烷化合物的合计量,优选为0.01倍摩尔~6倍摩尔,更优选为1倍摩尔~6倍摩尔。
硅烷化合物的水解·缩合反应时使用的溶媒优选为使用甲醇、乙醇、异丙醇、丁醇等醇类,水以及这些溶媒的混合溶媒等,但并不限定于此。反应中使用的溶媒优选为包含10质量%以上的水。更优选为水的含有比例为50质量%以上,进而优选为80质量%以上。相对于反应中使用的硅烷化合物的合计100质量份,溶媒的使用比例优选为10质量份~10,000质量份,更优选为50质量份~1,000质量份。
所述的水解·缩合反应优选为:首先,在包含催化剂及水的溶媒中混合硅烷化合物,进行6小时~24小时反应后,在开放体系中以50℃~70℃进行加热,使溶媒蒸发而实施。加热时间优选为设为0.5小时~24小时,更优选为设为1小时~12小时。在加热中,也可将混合液搅拌。另外,反应结束后,优选为进行使用离子交换树脂来转变分子内的反荷离子的处理。然后,视需要利用无水硫酸钙、分子筛等干燥剂将反应液干燥后,去除溶媒,借此可获得具有所述式(1)所表示的梯型结构的倍半硅氧烷。方法1中,优选为在至少满足条件1及条件2的条件下进行聚合,更优选为在满足所述三个条件的条件下进行聚合。
方法2中,通过使包含所述式(3)所表示的化合物的硅烷化合物进行水解·缩合而进行。所述式(3)中,R4的一价烃基优选为碳数1~10,更优选为甲基或乙基。R6优选为单键或碳数1~3。X4优选为氢原子或甲基。
作为所述式(3)所表示的化合物的具体例,例如可列举:对苯乙烯基三甲氧基硅烷、对苯乙烯基三乙氧基硅烷、4-乙烯基联苯三甲氧基硅烷、4-乙烯基环己基三甲氧基硅烷、4-环氧基苯基三甲氧基硅烷等。其中,优选为对苯乙烯基三甲氧基硅烷或者对苯乙烯基三乙氧基硅烷。此外,所述式(3)所表示的化合物可单独使用一种,也可将两种以上组合使用。
方法2的水解·缩合反应优选为在催化剂及溶媒的存在下进行。反应中使用的催化剂例如可列举碳酸钾、碳酸氢钠等。相对于硅烷化合物的总量,催化剂的使用比例优选为0.01倍摩尔~6倍摩尔。反应中使用的溶媒例如可列举:乙二醇二甲醚、乙二醇二乙醚、四氢呋喃、二恶烷等醚类,甲醇、乙醇、异丙醇、丁醇等醇类,水、以及这些溶媒的两种以上的混合溶媒等,但并不限定于此。关于水的含有比例,应用方法1的说明。反应温度优选为设为10℃~120℃,更优选为设为30℃~80℃。反应时间为0.1小时~48小时,优选为1小时~12小时。此外,方法2的反应时,也可并用所述式(3)所表示的化合物以外的其他的硅烷化合物作为硅烷化合物。相对于反应中使用的硅烷化合物的总量,所述其他的硅烷化合物的调配比例优选为设为70摩尔%以下,更优选为设为50摩尔%以下。
对于通过所述的水解·缩合反应而获得的倍半硅氧烷[L],利用凝胶渗透色谱法(GPC)来测定的聚苯乙烯换算的重量平均分子量(Mw)优选为在5,000~50,000的范围内。若Mw小于5,000,则存在对基板的涂布性劣化的倾向。另外,在利用液晶滴注方式(ODF(One Drop Filling)方式)来制造液晶显示元件的情况下,担忧产生因聚合体在液晶层中的流出而引起的显示不均(ODF不均)。另一方面,若Mw超过50,000,则聚合体容易凝胶化,液晶取向剂的保存稳定性劣化。就所述的观点而言,通过所述的水解·缩合反应而获得的倍半硅氧烷[L]的Mw的下限值优选为6,000以上,特别优选为8,000以上。Mw的上限值优选为45,000以下,更优选为40,000以下。另外,由重量平均分子量(Mw)与数量平均分子量(Mn)的比所表示的分子量分布(Mw/Mn)优选为在1.0~3.8的范围内,更优选为在1.0~3.3的范围内,进而优选为在1.0~3.0的范围内。
倍半硅氧烷[L]中的由下述式(4)所表示的部分结构(T3单元)而来的29Si-NMR光谱的峰值的积分比(以下也称为“T3峰值积分比”)优选为70%~99%。更优选为75%~99%,进而优选为80%~99%。此外,T3峰值积分比是表示含硅的聚合物中的T3单元(RISiO3/2)的含有比例的值,值越大,是指T3单元(RISiO3/2)的含有比例越高。
[化9]
(式(4)中,Ra为碳数1以上的一价有机基,“*”表示与硅原子的结合键)
作为倍半硅氧烷[L],在获得在所述式(1)所表示的结构中的E1、E2中具有选自由具有预倾角表现能力的基团、光取向性基及交联性基所组成的群组中的至少一种基团(以下也称为“功能性官能基”)的侧链修饰型聚合体的情况下,其合成方法并无特别限定,可通过将有机化学的常法适宜组合来进行。例如可列举:[I]对于特定硅烷化合物以及其他的硅烷化合物的至少任一者,使用具有功能性官能基的硅烷化合物进行聚合的方法;[II]使通过所述的水解·缩合反应而获得的倍半硅氧烷[L]所具有的官能基、与具有会与该官能基进行反应的基团以及功能性官能基的反应性化合物进行反应的方法等。
所述[I]的方法中,相对于倍半硅氧烷[L]的合成中使用的硅烷化合物的合计,具有功能性官能基的硅烷化合物的使用比例优选为设为0.5摩尔%~50摩尔%,更优选为设为1摩尔%~30摩尔%。此外,具有功能性官能基的硅烷化合物可单独使用一种或者将两种以上组合使用。
作为所述[II]的方法的具体例,例如可列举:使用具有功能性官能基的酰氯作为反应性化合物,在碱(例如氢氧化钠或吡啶、三乙胺等)的存在下与具有氨基或酚性羟基的倍半硅氧烷[L]进行反应的方法;使用具有功能性官能基的含氨基的化合物或者含羟基的化合物作为反应性化合物,且使用适当的缩合剂(例如:1-乙基-3-(3-二甲基氨基丙基)碳二酰亚胺(1-ethyl-3-(3-dimethylaminopropyl)carbodiimide,EDC)等),与具有羧基的倍半硅氧烷[L]进行反应的方法;使用具有功能性官能基的含环氧基的化合物作为反应性化合物,在层间转移催化剂(例如:溴化四甲基铵(tetramethyl ammonium bromide,TMAB)等)的存在下与具有羧基的倍半硅氧烷[L]进行反应的方法;使用具有功能性官能基的羧酸作为反应性化合物,在碱的存在下与具有苄基卤结构的倍半硅氧烷[L]进行反应的方法;使用具有功能性官能基的硫醇或胺作为反应性化合物,在自由基起始剂的存在下与具有苯乙烯基或烯基等的不饱和双键的倍半硅氧烷[L]进行反应的方法等。
反应性化合物若具有与倍半硅氧烷[L]所具有的官能基进行反应的基团以及功能性官能基,则其余的结构并无特别限定。作为反应性化合物的具体例,例如可列举下述式(r-1)~式(r-22)各自所表示的化合物等,但并不限定于这些化合物。
[化10]
[化11]
(式(r-1)~式(r-22)中,R15为碳数1~8的烷基或氟烷基,R16为氢原子或甲基,R17为烷基、环己基或苯基;Y1为单键或二价连结基,Z1为羧基、环氧基、-COC1、氨基或硫醇基,X5为羟基或氯原子;j为0~12的整数,h为1~20的整数;k为1~5的整数)
具有官能基的倍半硅氧烷与反应性化合物的反应时,就将足够量的功能性官能基导入至倍半硅氧烷的侧链上的观点而言,相对于倍半硅氧烷所具有的硅原子的合计,反应性化合物的使用比例优选为设为1摩尔%~90摩尔%,更优选为设为5摩尔%~50摩尔%。此外,反应性化合物可单独使用一种或者将两种以上组合使用。
关于在侧链上具有功能性官能基的倍半硅氧烷[L],利用GPC来测定的聚苯乙烯换算的重量平均分子量(Mw)优选为在6,000~60,000的范围内。Mw的下限值更优选为8,000以上,进而优选为10,000以上,特别优选为12,000以上。另外,Mw的上限值更优选为50,000以下,进而优选为45,000以下。分子量分布(Mw/Mn)优选为在1.0~3.8的范围内,更优选为在1.0~3.3的范围内,进而优选为在1.0~3.0的范围内。
液晶取向剂中所含有的倍半硅氧烷[L]在以将其溶解而成的反应溶液的形式来获得的情况下,可直接提供给液晶取向剂的制备,也可将反应溶液中所含的倍半硅氧烷[L]分离后再提供给液晶取向剂的制备。另外,也可将倍半硅氧烷[L]纯化后再提供给液晶取向剂的制备。倍半硅氧烷[L]的分离及纯化可依据公知的方法来进行。
依据包含所述倍半硅氧烷[L]的液晶取向剂,在利用残存于末端的硅烷醇基而获得倍半硅氧烷的交联密度充分高的液晶取向膜的方面优选。即,若液晶取向膜中的倍半硅氧烷的交联密度不充分,则在形成液晶单元的情况下,担忧由于倍半硅氧烷流入液晶中而使显示品质下降。就该方面而言,通过将倍半硅氧烷[L]用于取向膜材料中,随着交联密度的提高,膜成分在液晶中的流入减少,可确保显示品质。
依据所述的合成方法,可获得满足下述的必要条件(A)~(C)的聚合体作为倍半硅氧烷[L]。通过将满足这些必要条件(A)~(C)的聚合体用作液晶取向剂的聚合体成分,在对基板的涂布性良好、且可获得液晶取向性及电气特性良好的液晶元件的方面优选。
(A)利用GPC来测定的重量平均分子量(Mw)为5000以上。
(B)利用GPC来测定的由重量平均分子量(Mw)与数量平均分子量(Mn)的比所表示的分子量分布(Mw/Mn)为3.8以下。
(C)由所述式(4)所表示的部分结构而来的29Si-NMR光谱的峰值的积分比为70%~99%。
就充分确保所述效果的方面而言,关于必要条件(A),Mw优选为g000以上,更优选为10000以上,进而优选为12000以上。另外,关于必要条件(B),分子量分布更优选为3.3以下,进而优选为3.0以下。必要条件(C)的峰值积分比更优选为75%~99%,进而优选为80%~99%。此外,关于含硅的聚合物,通过T3峰值积分比大于T2峰值积分比(例如由所述式(4)所表示的部分结构而来的29Si-NMR光谱的峰值的积分比为70%以上),可溶于水中,重量平均分子量Mw大(例如Mw≥5000),且分子量分布(Mw/Mn)狭窄(例如分子量分布≤3.8),能够判断出所获得的含硅的聚合物的主骨架为梯型结构。倍半硅氧烷[L]可仅由所述式(1)所表示的梯型结构所构成,也可在一部分中包含无规型或笼型的结构。
<其他成分>
本发明的液晶取向剂含有如上所述的倍半硅氧烷[L],但也可视需要而含有其他成分。
所述其他成分之一可列举倍半硅氧烷[L]以外的其他聚合体。其他聚合体例如可列举:聚酰亚胺、聚酰胺酸、聚酰胺酸酯、不具有所述式(1)所表示的结构的聚硅氧烷(例如:笼型、无规型、双层结构(double-decker)型的聚硅氧烷)、聚酯、聚酰胺、纤维素衍生物、聚苯并恶唑前体、聚苯并恶唑、聚缩醛、聚苯乙烯衍生物、聚(苯乙烯-苯基顺丁烯二酰亚胺)衍生物、将聚(甲基)丙烯酸酯等作为主骨架的聚合体。此外,(甲基)丙烯酸酯是指包含丙烯酸酯及甲基丙烯酸酯。其他聚合体优选为这些聚合体中的选自由聚酰胺酸、聚酰亚胺及聚酰胺酸酯所组成的群组中的至少一种聚合体(以下也称为“聚合体[P]”)。
[聚酰胺酸1
作为聚合体[P]的聚酰胺酸例如可通过使四羧酸二酐与二胺进行反应而获得。
聚酰胺酸的合成中使用的四羧酸二酐可使用公知的四羧酸二酐。作为这些四羧酸二酐的具体例,脂肪族四羧酸二酐例如可列举:1,2,3,4-丁烷四羧酸二酐等;
脂环式四羧酸二酐例如可列举:1,2,3,4-环丁烷四羧酸二酐、2,3,5-三羧基环戊基乙酸二酐、1,3,3a,4,5,9b-六氢-5-(四氢-2,5-二氧代-3-呋喃基)-萘并[1,2-c]呋喃-1,3-二酮、1,3,3a,4,5,9b-六氢-8-甲基-5-(四氢-2,5-二氧代-3-呋喃基)-萘并[1,2-c]呋喃-1,3-二酮、2,4,6,8-四羧基双环[3.3.0]辛烷-2∶4,6∶8-二酐、环己烷四羧酸二酐等;芳香族四羧酸二酐例如可列举均苯四甲酸二酐等;除此以外,还可使用日本专利特开2010-97188号公报中记载的四羧酸二酐。此外,四羧酸二酐可单独使用一种或者将两种以上组合使用。
聚酰胺酸的合成中使用的二胺可使用公知的化合物。作为二胺的具体例,脂肪族二胺例如可列举:间苯二甲胺、1,3-丙二胺、四亚甲基二胺、六亚甲基二胺等;脂环式二胺例如可列举:1,4-二氨基环己烷、4,4′-亚甲基双(环己基胺)等;
芳香族二胺例如可列举:对苯二胺、4,4′-二氨基二苯基甲烷、4,4′-二氨基-2,2′-二甲基联苯、4,4′-二氨基-2,2′-双(三氟甲基)联苯、4,4′-二氨基二苯基醚、4-氨基苯基-4′-氨基苯甲酸酯、1,3-双(4-氨基苯氧基)丙烷、2,2-双[4-(4-氨基苯氧基)苯基]六氟丙烷、4,4′-(对亚苯基二亚异丙基)双苯胺、1,4-双(4-氨基苯氧基)苯、4,4′-双(4-氨基苯氧基)联苯、N,N′-双(4-氨基苯基)-联苯胺、1,4-双-(4-氨基苯基)-哌嗪、1-(4-氨基苯基)-2,3-二氢-1,3,3-三甲基-1H-茚-5-胺、3,5-二氨基苯甲酸、胆甾烷氧基-3,5-二氨基苯、3,5-二氨基苯甲酸胆甾烷基酯等;二氨基有机硅氧烷例如可列举:1,3-双(3-氨基丙基)-四甲基二硅氧烷等;除此以外,还可使用日本专利特开2010-97188号公报中记载的二胺。此外,这些二胺可单独使用一种或者将两种以上组合使用。
聚酰胺酸可通过使四羧酸二酐与二胺,视需要与分子量调整剂(例如:酸单酐、单胺化合物、单异氰酸酯化合物等)一起进行反应而获得。提供给聚酰胺酸的合成反应的四羧酸二酐与二胺的使用比例优选为相对于二胺的氨基1当量,四羧酸二酐的酸酐基成为0.2当量~2当量的比例。
聚酰胺酸的合成反应优选为在有机溶媒中进行。此时的反应温度优选为-20℃~150℃,反应时间优选为0.1小时~24小时。作为有机溶媒,优选为使用选自由N-甲基-2-吡咯烷酮、N,N-二甲基乙酰胺、N,N-二甲基甲酰胺、二甲基亚砜、γ-丁内酯、四甲基脲、六甲基磷酰三胺、间甲酚、二甲酚及卤化苯酚所组成的群组中的一种以上作为溶媒,或者使用这些溶媒的一种以上与选自由醇、酮、酯、醚、卤化烃及烃所组成的群组中的一种以上的混合物。将聚酰胺酸溶解而成的反应溶液可直接提供给液晶取向剂的制备,也可将反应溶液中所含的聚酰胺酸分离后再提供给液晶取向剂的制备。
[聚酰亚胺]
作为聚合体[P]的聚酰亚胺可通过将以所述方式合成的聚酰胺酸进行脱水闭环,加以酰亚胺化而获得。聚酰亚胺可为将聚酰胺酸所具有的酰胺酸结构全部进行脱水闭环而成的完全酰亚胺化物,也可为酰胺酸结构与酰亚胺环结构并存的部分酰亚胺化物。聚酰亚胺的酰亚胺化率优选为30%以上,更优选为40%~99%。该酰亚胺化率是相对于聚酰亚胺的酰胺酸结构的数量与酰亚胺环结构的数量的合计,以百分率来表示酰亚胺环结构的数量所占的比例。此处,酰亚胺环的一部分也可为异酰亚胺环。
聚酰胺酸的脱水闭环优选为利用如下方法来进行:将聚酰胺酸溶解于有机溶媒中,在该溶液中添加脱水剂及脱水闭环催化剂,视需要进行加热。脱水剂例如可使用乙酸酐、丙酸酐、三氟乙酸酐等酸酐。相对于聚酰胺酸的酰胺酸结构的1摩尔,脱水剂的使用量优选为设为0.01摩尔~20摩尔。脱水闭环催化剂例如可使用吡啶、三甲吡啶、二甲吡啶、三乙胺等三级胺。相对于所使用的脱水剂1摩尔,脱水闭环催化剂的使用量优选为设为0.01摩尔~10摩尔。脱水闭环反应中使用的有机溶媒可列举作为聚酰胺酸的合成中所使用者而例示的有机溶媒。脱水闭环反应的反应温度优选为0℃~180℃,反应时间优选为1.0小时~120小时。含有聚酰亚胺的反应溶液可直接提供给液晶取向剂的制备,也可将聚酰亚胺分离后再提供给液晶取向剂的制备。除此以外,聚酰亚胺也可通过聚酰胺酸酯的酰亚胺化而获得。
[聚酰胺酸酯]
作为聚合体[P]的聚酰胺酸酯例如可利用以下方法来获得:[I]使通过所述合成反应而获得的聚酰胺酸与酯化剂进行反应的方法;[II]使四羧酸二酯与二胺进行反应的方法;[III]使四羧酸二酯二卤化物与二胺进行反应的方法等。液晶取向剂中所含有的聚酰胺酸酯可仅具有酰胺酸酯结构,也可以是酰胺酸结构与酰胺酸酯结构并存的部分酯化物。此外,将聚酰胺酸酯溶解而成的反应溶液可直接提供给液晶取向剂的制备,也可将反应溶液中所含的聚酰胺酸酯分离后再提供给液晶取向剂的制备。
以所述方式获得的聚酰胺酸、聚酰胺酸酯以及聚酰亚胺优选为当将其制成浓度为10质量%的溶液时,具有10mpa·s~800mpa·s的溶液粘度,更优选为具有15mpa·s~500mpa·s的溶液粘度。此外,所述聚合体的溶液粘度(mpa·s)是对使用该聚合体的良溶媒(例如γ-丁内酯、N-甲基-2-吡咯烷酮等)来制备的浓度为10质量%的聚合体溶液,使用E型旋转粘度计在25℃下测定而得的值。对于液晶取向剂中所含有的聚酰胺酸、聚酰胺酸酯以及聚酰亚胺,利用凝胶渗透色谱法(GPC)来测定的聚苯乙烯换算的重量平均分子量优选为500~100,000,更优选为1,000~50,000。
调配于液晶取向剂中的聚合体成分的优选形态可列举以下的(i)及(ii)。
(i)单独含有倍半硅氧烷[L]作为聚合体成分的形态。
(ii)含有倍半硅氧烷[L]及聚合体[P]作为聚合体成分的形态。
在所述(ii)的情况下,就适宜获得由倍半硅氧烷[L]的调配所带来的涂布性及可靠性的改善效果的观点而言,倍半硅氧烷[L]与聚合体[P]的调配比例([L]:[P])优选为以质量比计而设为1∶99~80∶20。更优选为10∶90~75∶25,进而优选为20∶80~70∶30。
此外,作为其他成分,所述以外的添加剂例如可列举:分子内具有至少一个环氧基的化合物、官能性硅烷化合物、分子内具有至少一个氧杂环丁基的化合物、抗氧化剂、表面活性剂、光增感剂、填充剂、消泡剂、分散剂、密合助剂、抗静电剂、流平剂、抗菌剂等。这些添加剂的调配比例可根据所使用的化合物,在不妨碍本发明效果的范围内适宜设定。
<溶剂>
本发明的液晶取向剂被制备成倍半硅氧烷[L]以及视需要使用的其他成分优选为在适当的溶媒中分散或者溶解而成的液状组合物。所使用的有机溶媒例如可列举:N-甲基-2-吡咯烷酮、γ-丁内酯、γ-丁内酰胺、N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、4-羟基-4-甲基-2-戊酮、乙二醇单甲醚、乳酸丁酯、乙酸丁酯、甲基甲氧基丙酸酯、乙基乙氧基丙酸酯、乙二醇甲醚、乙二醇乙醚、乙二醇-正丙醚、乙二醇-异丙醚、乙二醇-正丁醚(丁基溶纤剂)、乙二醇二甲醚、乙二醇乙醚乙酸酯、二乙二醇二甲醚、二乙二醇二乙醚、二乙二醇单甲醚、二乙二醇单乙醚、二乙二醇单甲醚乙酸酯、二乙二醇单乙醚乙酸酯、二异丁基酮、丙酸异戊酯、异丁酸异戊酯、二异戊基醚、碳酸亚乙酯、碳酸亚丙酯等。这些有机溶媒可单独使用或者将两种以上混合使用。
液晶取向剂中的固体成分浓度(液晶取向剂的溶媒以外的成分的合计质量在液晶取向剂的总质量中所占的比例)可考虑到粘性、挥发性等来适宜选择,优选为1质量%~10质量%的范围。即,本发明的液晶取向剂是通过以后述方式涂布于基板表面,优选为进行加热,从而形成作为液晶取向膜的涂膜或者成为液晶取向膜的涂膜。此时,在固体成分浓度小于1质量%的情况下,涂膜的膜厚变得过小而难以获得良好的液晶取向膜。另一方面,在固体成分浓度超过10质量%的情况下,涂膜的膜厚变得过大而难以获得良好的液晶取向膜,另外,存在液晶取向剂的粘性增大而涂布性下降的倾向。制备本发明的液晶取向剂时的温度优选为10℃~50℃。
[液晶取向膜及液晶元件]
本发明的液晶取向膜是利用以所述方式制备的液晶取向剂来形成。另外,本发明的液晶元件包括使用所述液晶取向剂而形成的液晶取向膜。液晶元件中的液晶的动作模式并无特别限定,可应用于扭转向列(Twisted Nematic,TN)型、超扭转向列(Super Twisted Nematic,STN)型、共面切换(In-Plane Switching,IPS)型、边缘场切换(Fringe Field Switching,FFS)型、垂直取向(Vertical Alignment,VA)型、多域垂直取向(Multidomain Vertical Alignment,MVA)型、聚合物稳定取向(Polymer sustained alignment,PSA)型等多种模式。
液晶元件可利用例如包含以下的步骤1~步骤3的方法来制造。步骤1根据所需的驱动模式而使用不同的基板。步骤2及步骤3在各模式中共通。
[步骤1:涂膜的形成]
首先,在基板上涂布本发明的液晶取向剂,继而对涂布面进行加热,借此在基板上形成涂膜。基板例如可使用:浮法玻璃、钠玻璃等玻璃;包含聚对苯二甲酸乙二酯、聚对苯二甲酸丁二酯、聚醚砜、聚碳酸酯、聚(脂环式烯烃)等塑料的透明基板。在制造TN型、STN型、VA型、MVA型或PSA型的液晶显示元件的情况下,使用两块设置有经图案化的透明导电膜的基板。在制造IPS型或FFS型液晶显示元件的情况下,使用设置有包含经图案化为梳齿型的透明导电膜或者金属膜的电极的基板、以及未设置电极的对向基板。设置于基板面的透明导电膜可使用包含氧化锡(SnO2)的奈塞(NESA)膜(美国PPG公司注册商标)、包含氧化铟-氧化锡(In2O3-SnO2)的ITO膜等。金属膜可使用包含例如铬等金属的膜。液晶取向剂在基板上的涂布优选为利用胶版印刷法、旋涂法、辊涂布机法或者喷墨印刷法来进行。
涂布液晶取向剂后,出于防止所涂布的取向剂的流挂等目的,优选为实施预加热(预烘烤)。预烘烤温度优选为30℃~200℃,预烘烤时间优选为0.25分钟~10分钟。然后,出于将溶剂完全去除的目的,另外,在液晶取向剂包含聚合体[P]的情况下,出于视需要将存在于聚合体[P]中的酰胺酸结构进行热酰亚胺化的目的,而实施煅烧(后烘烤)步骤。此时的煅烧温度(后烘烤温度)优选为80℃~300℃。后烘烤时间优选为5分钟~200分钟。以所述方式形成的膜的膜厚优选为0.001μm~1μm。于基板上涂布液晶取向剂后,将有机溶媒去除,借此形成成为液晶取向膜的涂膜。
[步骤2:取向能力赋予处理]
在制造TN型、STN型、IPS型或FFS型的液晶显示元件的情况下,实施对所述步骤1中形成的涂膜赋予液晶取向能力的处理。借此,液晶分子的取向能力被赋予至涂膜而成为液晶取向膜。取向能力赋予处理例如可列举以下处理等:摩擦处理,利用卷绕有包含尼龙、人造丝、棉等纤维的布的辊,将涂膜向一定方向擦拭;光取向处理,对涂膜照射偏光或非偏光的放射线。
在通过光取向处理对涂膜赋予液晶取向能力的情况下,对涂膜照射的放射线例如可使用包含150nm~800nm波长的光的紫外线以及可见光线,可为偏光,也可为非偏光。所使用的光源例如可使用:低压水银灯、高压水银灯、氘灯、金属卤化物灯、氩共振灯、氙灯、准分子激光等。放射线的照射量优选为100J/m2~50,000J/m2,更优选为300J/m2~20,000J/m2。另外,为了提高反应性,可一边对涂膜加温一边对涂膜进行光照射。适合于VA型液晶显示元件的液晶取向膜也可适合用于聚合物稳定取向(Polymer sustained alignment,PSA)型的液晶显示元件。
[步骤3:液晶单元的构筑]
准备两块以所述方式形成有液晶取向膜的基板,在对向配置的两块基板间配置液晶,借此制造液晶单元。制造液晶单元的方法例如可列举:(1)以各个液晶取向膜对向的方式经由单元间隙而将两块基板对向配置,使用密封剂将两块基板的周边部贴合,在由基板表面及密封剂所划分的单元间隙内注入填充液晶后,将注入孔密封的方法;(2)液晶滴注方式(ODF方式)等。密封剂例如可使用含有硬化剂以及作为间隔物的氧化铝球的环氧树脂等。液晶可列举向列液晶及碟状液晶,其中优选为向列液晶。另外,也可以在这些液晶中添加例如胆甾醇型液晶、手性剂、铁电性液晶等来使用。
接着,可通过视需要在液晶单元的外侧表面贴合偏光板来获得本发明的液晶元件。贴合于液晶单元的外表面的偏光板可列举:以乙酸纤维素保护膜夹持被称为“H膜”的偏光膜而成的偏光板或者包含H膜其本身的偏光板,所述“H膜”是一边使聚乙烯醇延伸取向一边吸收碘而成。
本发明的液晶元件可有效地应用于多种装置,例如可用于:钟表、便携型游戏机、文字处理器(word processor)、笔记型个人计算机(note type personal computer)、汽车导航系统(car navigation system)、摄像机(camcorder)、个人数字助理(Personal Digital Assistant,PDA)、数码照相机(digital camera)、手机、智能手机、各种监视器、液晶电视等显示装置,或调光膜等。另外,使用本发明的液晶取向剂而形成的液晶元件也可应用于相位差膜。
[实施例]
以下,通过实施例,对本发明进一步进行具体说明,但本发明并不限定于这些实施例。
以下例中,聚合体的重量平均分子量、聚酰亚胺的酰亚胺化率、聚合体溶液的溶液粘度、环氧当量、以及T3峰值积分比是利用以下方法来测定。此外,以下例中的“份”及“%”只要无特别说明,则为质量基准。
[聚合体的重量平均分子量及数量平均分子量]:利用以下条件下的GPC来测定的聚苯乙烯换算值。
管柱:东曹(股)制造,TSKgelGRCXLII
溶剂:四氢呋喃
温度:40℃
压力:68kgf/cm2
[聚酰亚胺的酰亚胺化率]:将含有聚酰亚胺的溶液投入至纯水中,将所获得的沉淀在室温下充分地减压干燥后,溶解于氘化二甲基亚砜中,将四甲基硅烷作为基准物质,在室温下测定1H-核磁共振(Nuclear Magnetic Resonance,NMR)。根据所获得的1H-NMR光谱,使用下述数式(1)来求出酰亚胺化率[%]。
酰亚胺化率[%]=(1-A1/A2×α)×100…(1)
(数式(1)中,A1是在化学位移10ppm附近出现的源自NH基的质子的峰值面积,A2是源自其他质子的峰值面积,α是其他质子相对于聚酰胺酸中的NH基的一个质子的个数比例)
[聚合体溶液的溶液粘度(mpa·s)]:对于使用既定的溶媒而制备成聚合体浓度10质量%的溶液,使用E型旋转粘度计在25℃下测定。
[环氧当量]:依据JIS C2105的“盐酸-甲基乙基酮法”来测定。
[T3峰值积分比]:通过29Si-NMR,以如下方式求出。将使固体成分浓度为20%的2g的聚硅氧烷溶液以及三(2,4-戊二酮基)铬(III)溶解于0.8g的氘代二甲基亚砜(dimethylsulfoxide-d6,重DMSO)中而成的作为样品,使用核磁共振装置阿旺斯(AVANCE)III400型NMR装置(布鲁克拜厄斯宾(Bruker BioSpin)公司)来进行所述样品的29Si-NMR测定。基于通过所述29Si-NMR测定而获得的各光谱的化学位移的差异,来求出与聚硅氧烷的下述式(T1)~式(T3)各自所表示的各结构的含有比例相当的各个信号的积分值。
[化12]
(式(T1)~式(T3)中,Ra为碳数1以上的一价有机基,“*”表示与硅原子的结合键)
<倍半硅氧烷[L]的合成>
以下述的程序来合成倍半硅氧烷[L]。此外,合成例1-1~合成例1-6及合成例1-8中使用方法1,合成例1-7中使用方法2。
[合成例1-1]
将作为条件1的特定硅烷化合物的4.43g(20mmol)的3-氨基丙基三乙氧基硅烷,混合于1.0mol/L的60mL盐酸水溶液(含有溶媒整体的96质量%的水)中,在室温下搅拌2小时。继而,在开放体系中以60℃~70℃进行加热,使溶媒蒸发而进行缩聚。接着,将产物在100℃下放置一晚后,溶解于300mL的水中,将所获得的水溶液进行冷冻干燥,借此获得4.24g(产率为94%)的白色粉末状的化合物(以下作为“聚合体(L-1A)”)。
继而,将所获得的聚合体(L-1A)的0.20g溶解于50mL的水中,通过填充有阴离子交换树脂(安博莱特(Amberlite)IRA-900)的管柱,进而输送作为条件2的催化剂的1N的NaOH水溶液。然后,将所获得的溶液冷冻干燥,借此获得1.63g(产率为74%)的具有下述式(s-1)所表示的部分结构的倍半硅氧烷(以下称为“聚合体(L-1)”)。所获得的聚合体(L-1)的Mw=13,000,Mn=6,400,Mw与Mn的比(Mw/Mn)所表示的分子量分布(PDI)=2.0,T3峰值积分比为89%,可溶于水。根据这些物性,确认聚合体(L-1)为梯型。
另外,根据将该聚合体溶液在20℃下静置了3天时的凝胶化的有无来评价保存稳定性。将未看到凝胶化的情况评价为保存稳定性“良好”,将看到凝胶化的情况评价为“不良”。其结果为,该聚合体溶液未凝胶化,保存稳定性为“良好”。
[化13]
[合成例1-2及合成例1-3]
除了将所使用的单体的种类及量变更为如下述表1所述以外,通过进行与合成例1-1相同的操作而分别获得梯型倍半硅氧烷(聚合体(L-2)及聚合体(L-3))。将所获得的聚合体的物性示于下述表2中。
[合成例1-4]
将作为条件1的特定硅烷化合物的4.35g的2-氰基乙基三乙氧基硅烷,混合于作为条件2的催化剂的5mL的浓度为8%的氢氧化钠水溶液中,搅拌13小时。将所获得的水溶液在开放体系中以50℃~60℃进行加热,使溶媒蒸发而进行缩聚。然后,将所生成的粉末状粗产物添加于加入有约100cm3的H+类型的阳离子交换树脂的100mL水中,在室温下搅拌3小时。将阳离子交换树脂过滤分离后,将所获得的水溶液以旋转蒸发器(rotary evaporator)来浓缩至20mL,然后进行冷冻干燥,借此获得1.95g(产率为70%)的具有下述式(s-4)所表示的部分结构的倍半硅氧烷(以下称为“聚合体(L-4)”)。所获得的聚合体(L-4)的Mw为8,000,Mn为4,000,分子量分布(PDI)为2.0,T3峰值积分比为86%,可溶于水。根据这些物性,确认聚合体(L-4)为梯型。另外,关于该聚合体溶液,以与合成例1-1相同的方式评价保存稳定性,结果为“良好”。
[化141
[合成例1-5]
除了将所使用的单体的种类及量变更为如下述表1所述以外,通过进行与合成例1-4相同的操作而获得梯型倍半硅氧烷(聚合体(L-5))。将所获得的聚合体的物性示于下述表2中。
[合成例1-6]
除了将所使用的单体的种类及量变更为如下述表1所述的方面、以及使用氢氧化钠水溶液与过氧化氢水溶液的混合溶液作为催化剂的方面以外,通过进行与合成例1-4相同的操作而获得梯型倍半硅氧烷(聚合体(L-6))。将所获得的聚合体的物性示于下述表2中。
[合成例1-7]
将作为所述式(3)所表示的化合物的9.00g的对苯乙烯基三甲氧基硅烷以及0.04g的碳酸钾,添加于四氢呋喃/水的混合溶媒7mL(质量比为3/2)中进行混合,在40℃下进行2小时聚合。继而,使沉淀的粘性固体溶解于15mL的二氯甲烷中,将所获得的液体缓缓地滴加于100mL的甲醇中。滴加后滤取所产生的固体,使其干燥,借此以产率77%获得作为倍半硅氧烷的聚合体(L-7)。所获得的聚合体(L-7)的Mw为9,700,Mn为4,800,分子量分布(PDI)为2.0,T3峰值积分比为92%,可溶于水。将聚合体(L-7)的29Si-NMR图表示于图1中。此外,所述式(4)所表示的部分结构中的Ra为苯乙烯基时的T3单元的峰值出现在-80ppm附近,T2单元的峰值出现在-70ppm附近,T1单元的峰值出现在-65ppm附近。根据图1的结果,聚合体(L-7)的T3单元的比例多于T2单元及T1单元。根据这些结果,确认聚合体(L-7)为梯型。
[合成例1-8]
在氮气环境下,在3.17g(0.025mol)的氯二甲氧基硅烷中添加15mL的二甲苯溶液,冷却至-15℃。继而,缓缓地滴加2.70g(0.025mol)的对苯二胺的丙酮溶液25ml,然后搅拌30分钟。搅拌后,依次滴加15mL的丙酮、20mL的二甲苯、6mL(10mol/L)的盐酸,在室温下搅拌4小时。然后,添加硫酸钠进行脱水,通过过滤来去除硫酸钠。在滤液中添加2滴浓硫酸,在室温下搅拌3小时后,在90℃下搅拌3小时。继而,以50ml的水进行分液洗涤,将有机层的溶媒蒸馏去除,借此以产率83%获得作为倍半硅氧烷的聚合体(L-8)。所获得的聚合体(L-8)的Mw为15,000,Mn为8,000,分子量分布(PDI)为1.9,T3峰值积分比为82%,可溶于水。根据以上的物性,确认聚合体(L-8)为梯型。
<其他聚合体(聚硅氧烷)的合成>
[比较合成例1-1]
使4.9g(20mmol)的2-(3,4-环氧基环己基)乙基三甲氧基硅烷溶解于50g的环戊酮中,缓缓滴加于将0.2g的三乙胺(triethylamine,TEA)及5.0g的水溶解于50g的环戊酮中而成的溶液中,进行混合,在55℃下搅拌2小时。利用蒸发器使溶媒蒸发,将产生的固体以丙酮进行洗涤,获得3.30g(产率93%)的作为白色粉末状化合物的笼型倍半硅氧烷(以下作为“聚合体(R-1)”)。所获得的聚合体(R-1)的Mw为3,500,Mn为2,100,分子量分布(PDI)为1.7,T3峰值积分比为94%。另外,将该聚合体溶液在20℃下静置3天后,未凝胶化,保存稳定性为良好。
[比较合成例1-2~比较合成例1-4]
除了将所使用的单体的种类及量变更为如下述表1所述以外,通过进行与比较合成例1-1相同的操作而分别获得作为笼型倍半硅氧烷的聚合体(R-2)~聚合体(R-4)。将所获得的聚合体的物性一并示于下述表1中。此外,对于聚合体(R-3)及聚合体(R-4),将聚合体溶液在20℃下静置3天后,观察到凝胶化,保存稳定性为不良。
[比较合成例1-5]
在具备回流管的500mL的四口反应烧瓶中,投入51.3g的己二醇、15.6g的丁基溶纤剂、66.0g的四乙氧基硅烷、以及6.9g的十二烷基三乙氧基硅烷而进行搅拌。向该溶液中,在室温下滴加预先将25.6g的己二醇、7.8g的丁基溶纤剂、30.0g的水以及作为催化剂的0.3g的草酸混合而成的草酸溶液,滴加结束后搅拌30分钟。然后,在溶液温度70℃下加热1小时后放置冷却。在该溶液中添加己二醇(hexyleneglycol,HG)、丁基溶纤剂(butyl eellosolve,BC)及N-甲基-2-吡咯烷酮(N-methyl-2-pyrrolidone,NMP),HG∶BC∶NMP=30∶50∶20(质量比),且以SiO2换算浓度成为3.5质量%的方式来制备,获得含有无规型聚硅氧烷(以下作为“聚合体(R-5)”)的溶液。所获得的聚合体(R-5)的Mw为9,000,Mn为2,650,分子量分布(PDI)为3.4,T3峰值积分比为约0%(表2)。
[比较合成例1-6]
除了代替1.0mol/L的盐酸水溶液而使用相同浓度的氢氧化钠水溶液的方面以外,通过进行与比较合成例1-1相同的操作而获得作为倍半硅氧烷的聚合体(R-6)。所获得的聚合体(R-6)的Mw为125,000,Mn为29,000,分子量分布(PDI)为4.3,T3峰值积分比为62%(表2),根据这些物性,确认聚合体(R-6)为无规型。该聚合物在一天后凝胶化。将所获得的聚合体的物性示于下述表2中。
[比较合成例1-7]
除了代替浓度为8%的氢氧化钠水溶液,而使用相同浓度的盐酸水溶液的方面以外,通过进行与比较合成例1-4相同的操作而获得作为倍半硅氧烷的聚合体(R-7)。所获得的聚合体(R-7)的Mw为60,000,Mn为15,000,分子量分布(PDI)为4.0,T3峰值积分比为94%(表2),根据这些物性,确认聚合体(R-7)为无规型。
[表1]
表1中,单体的数值表示相对于反应中使用的单体的合计而言的各化合物的使用比例(摩尔%)。
<单体>
MS3-1:2-(3,4-环氧基环己基)乙基三甲氧基硅烷
MS3-2:3-甲基丙烯酰氧基丙基三甲氧基硅烷
MS3-3:十二烷基三乙氧基硅烷
MS3-4:3-氨基丙基三乙氧基硅烷
MS3-5:2-氰基乙基三乙氧基硅烷
MS3-6:4-(氯甲基)苯基三甲氧基硅烷
MS3-7:对苯乙烯基三甲氧基硅烷
MS3-8:3-巯基丙基三乙氧基硅烷
MS3-9:氯二甲氧基硅烷
MS4-1:四乙氧基硅烷
[表2]
<侧链修饰型的倍半硅氧烷[L]的合成>
[合成例2-1]
在1.02g(3.7mmol)的4-(4-正戊基环己基)苯甲酸中混合10mL的亚硫酰氯,在60℃下加温1小时。然后,通过减压蒸馏去除而去除体系中的亚硫酰氯,获得白色的酰氯。使其溶解于20g的四氢呋喃中。继而,使1.63g的聚合体(L-1)溶解于15ml的水及15mL的四氢呋喃的混合溶媒中,在该溶液中加入0.45g(4.4mmol)的三乙胺以及0.05g的溴化四丁基铵,进行搅拌。将其置于冰浴冷却下,将体系中的内温设为5℃以下。在该溶液中,花一小时滴加刚才的酰氯溶液,在冰浴下搅拌4小时。
继而,在所获得的反应液中添加100mL的环戊酮,进行分液萃取。然后,添加150mL的N-甲基-2-吡咯烷酮,以蒸发器使其浓缩,获得含有侧链修饰型聚合体(L-11)的聚合体溶液。所获得的聚合体(L-11)的Mw为19,000,Mn为9,600,分子量分布(PDI)为2.0,T3峰值积分比为91%。另外,保存稳定性的评价为“良好”。
[合成例2-2及合成例2-3]
除了将所使用的羧酸的种类及量变更为如下述表3所述以外,通过进行与合成例2-1相同的操作而分别获得聚合体(L-12)及聚合体(L-13)。将所获得的聚合体的物性示于下述表4中。
[合成例2-4]
使作为反应性化合物(侧链成分)的0.69g(2.8mmol)的4-(4-正戊基环己基)苯胺、以及1.95g的聚合体(L-4),溶解于20mL的N-甲基-2-吡咯烷酮以及40mL的环戊酮的混合溶媒中。将其置于冰浴冷却下,将体系中的内温设为5℃以下。加入0.81g(4.2mmol)的1-乙基-3-(3-二甲基氨基丙基)碳二酰亚胺盐酸盐、以及0.1g(催化剂量)的二甲基氨基吡啶,在冰浴冷却下进行6小时反应。继而,在所获得的反应液中添加100mL的环戊酮,进行分液萃取。然后,在有机层中添加100mL的N-甲基-2-吡咯烷酮,以蒸发器进行浓缩,获得含有侧链修饰型聚合体(L-14)的聚合体溶液。
[合成例2-5及合成例2-6]
除了将所使用的反应性化合物的种类及量变更为如下述表3所述以外,通过进行与合成例2-4相同的操作而分别获得聚合体(L-15)及聚合体(L-16)。将所获得的聚合体的物性示于下述表4中。
[合成例2-7]
使作为反应性化合物(侧链成分)的1.01g(3.7mmol)的4-(4-正戊基环己基)苯甲酸溶解于30mL的四氢呋喃(tetrahydrofuran,THF)中,添加2.56g(18.5mmol)的碳酸钾。继而,加入2.51g的聚合体(L-5),在室温下搅拌3小时。搅拌后,通过过滤而去除固体。继而,在所获得的滤液中添加100mL的环戊酮,进行分液萃取。然后,在有机层中添加100mL的N-甲基-2-吡咯烷酮,以蒸发器进行浓缩,获得含有侧链修饰型聚合体(L-17)的聚合体溶液。将所获得的聚合体的物性示于下述表4中。
[合成例2-8]
使1.01g(3.02mmol)的下述式(mc-7)所表示的化合物溶解于30mL的甲苯中,继而添加1.98g的聚合体(L-7)以及9.85mg的偶氮双异丁腈,在70℃下搅拌3小时。反应后,添加100mL的N-甲基-2-吡咯烷酮,以蒸发器进行浓缩,获得含有侧链修饰型聚合体(L-18)的聚合体溶液。将所获得的聚合体的物性示于下述表4中。
[合成例2-9]
在5.15g的聚合体(L-8)中添加30mL的甲苯,使其溶解。继而,依次添加6.21g(50mol)的1,2-环氧基-4-乙烯基环己烷、0.1g的六氯铂(IV)酸,在80℃下进行2小时反应。反应后,进行过滤而去除催化剂。在滤液中添加100mL的环戊酮进行浓缩,借此获得30g的固体成分浓度为30质量%的环戊酮溶液。在所获得的溶液中添加4.12g(15mmol)的4-(4-正戊基环己基)苯甲酸、0.5g的溴化四丁基铵,在110℃下搅拌4小时。搅拌后,添加100mL的水,进行分液纯化。继而,在有机层中添加30mL的丁基溶纤剂,进行浓缩,借此获得含有侧链修饰型聚合体(L-19)的聚合体溶液。将所获得的聚合体的物性示于下述表4中。
<其他聚合体(侧链修饰型倍半硅氧烷)的合成>
[比较合成例2-1]
在100mL的三口烧瓶中,投入3.3g的聚合体(R-1)、50g的甲基异丁基酮、1.02g的4-(4-正戊基环己基)苯甲酸、50mg的2,6-二-叔丁基-4-甲氧基苯酚、以及0.6g的商品名“UCAT 18X”(三亚普罗(San-Apro)(股)制造,环氧化合物的硬化促进剂),在90℃下进行48小时反应。反应结束后,在反应混合物中添加甲醇而生成沉淀,对将该沉淀物溶解于乙酸乙酯中而获得的溶液水洗3次后,将溶剂蒸馏去除,借此获得3.1g的侧链修饰型倍半硅氧烷(聚合体(R-11))的白色粉末。该聚合体(R-11)的Mw为4,500,Mn为2,800,分子量分布(PDI)为1.6,T3峰值积分比为94%。另外,保存稳定性的评价为“良好”。
[比较合成例2-2]
除了将所使用的羧酸的种类及量变更为如下述表3所述以外,通过进行与比较合成例2-1相同的操作而获得聚合体(R-12)。将所获得的聚合体的物性示于下述表4中。
[表3]
表3中,反应性化合物的数值表示相对于反应中使用的聚合体所具有的硅原子而言的反应性化合物的使用比例(摩尔%)。表3中的反应性化合物的略称分别为以下的含义。MC-5~MC-7为以下的结构。
<反应性化合物>
MC-1:4-(4-正戊基环己基)苯甲酸
MC-2:3,5-双(甲基丙烯酰氧基)苯甲酸
MC-3:4-苯氧基肉桂酸
MC-4:4-(4-正戊基环己基)苯胺
MC-5:下述式(mc-5)所表示的化合物
MC-6:下述式(mc-6)所表示的化合物
MC-7:下述式(mc-7)所表示的化合物
[化15]
[表4]
<其他聚合体(聚合体[P])的合成>
[合成例3-1]
将19.61g(0.1摩尔)的1,2,3,4-环丁烷四羧酸二酐及21.23g(0.1摩尔)的4,4′-二氨基-2,2′-二甲基联苯溶解于367.6g的N-甲基-2-吡咯烷酮中,在室温下进行6小时反应,获得包含聚酰胺酸(PA-1)的溶液。
[合成例3-2]
将作为酸二酐的21.8g(0.1摩尔)的均苯四甲酸二酐及19.6g(0.1摩尔)的1,2,3,4-环丁烷四羧酸二酐、以及作为二胺的40g(0.2摩尔)的4,4′-二氨基二苯基醚溶解于458g的N-甲基-2-吡咯烷酮中,在40℃下进行3小时反应后,追加356g的N-甲基-2-吡咯烷酮,借此获得包含聚酰胺酸(PA-2)的溶液。
[合成例3-3]
将19.61g(0.1摩尔)的1,2,3,4-环丁烷四羧酸二酐及22.8g(0.1摩尔)的4-氨基苯基-4′-氨基苯甲酸酯,溶解于382g的N-甲基-2-吡咯烷酮中,在室温下进行12小时反应,获得包含聚酰胺酸(PA-3)的溶液。
[合成例3-4]
将22.4g(0.1摩尔)的2,3,5-三羧基环戊基乙酸二酐及10.81g(0.1摩尔)的对苯二胺,溶解于329.3g的N-甲基-2-吡咯烷酮中,在60℃下进行6小时反应。继而,将反应混合物注入至大量过剩的甲醇中,使反应产物沉淀。将沉淀物以甲醇进行洗涤,在减压下以40℃干燥15小时,借此获得32g的聚酰胺酸(PA-4)。
秤取17.5g的所述合成中获得的聚酰胺酸(PA-4),在其中添加232.5g的N-甲基-2-吡咯烷酮、3.8g的吡啶及4.9g的乙酸酐,在120℃下进行4小时反应而进行酰亚胺化。继而,将反应混合液注入至大量过剩的甲醇中,使反应产物沉淀。将沉淀物以甲醇进行洗涤,在减压下干燥15小时,借此获得15g的聚酰亚胺的粉末。使其溶解于135g的N-甲基-2-吡咯烷酮中,获得10质量%溶液的聚酰亚胺(PI-1)。
<液晶取向剂及液晶显示元件的评价>
[实施例1]
(1)液晶取向剂的制备
将含有聚酰胺酸(PA-1)的溶液,秤取换算为其中所含有的聚酰胺酸(PA-1)而相当于100质量份的量,在其中添加100质量份的所述合成例2-3中获得的聚合体(L-13),进而添加N-甲基-2-吡咯烷酮(NMP)以及丁基溶纤剂(BC),制成溶媒组成为NMP∶BC=80∶20(质量比)、固体成分浓度为3.5质量%的溶液。将该溶液以孔径为1μm的过滤器进行过滤,借此制备液晶取向剂(A-1)。
(2)印刷性的评价
使用液晶取向膜印刷机(日本写真印刷(股)制造),将所述(1)中制备的液晶取向剂(A-1)涂布于带有包含ITO膜的透明电极的玻璃基板的透明电极面上,在80℃的加热板上加热(预烘烤)1分钟而去除溶媒后,在230℃的加热板上加热(后烘烤)10分钟,形成平均膜厚为0.06μm的涂膜。利用倍率为20倍的显微镜对该涂膜进行观察,来调查膜厚不均、橘皮缺陷不均以及线状不均的有无。将无膜厚不均、涂布面内均匀者评价为印刷性“良好(○)”,将观察到橘皮缺陷不均的情况评价为印刷性“可(△)”,将观察到橘皮缺陷不均及线状不均的情况评价为印刷性“不良(×)”。本实施例中,未观察到膜厚不均,另外,涂布面内均匀,印刷性为“良好(○)”。
(3)残膜率的评价
使用旋转器,将所述(1)中制备的液晶取向剂(A-1)涂布于玻璃基板上,在80℃的加热板上进行1分钟预烘烤后,在将库内进行了氮气置换的230℃的烘箱中进行1小时加热(后烘烤),借此形成涂膜。继而,在后烘烤后的膜上,通过旋涂来涂布N-甲基-2-吡咯烷酮(NMP)。利用扫描型电子显微镜来测定旋涂NMP之前的膜厚D1[μm]以及涂布后的膜厚D2[μm],利用下述数式(2)来算出残膜率(%)。
残膜率(%)=[(D1-D2)/D1]×100…(2)
残膜率越高,表示倍半硅氧烷的交联密度越高。另一方面,若倍半硅氧烷的交联密度低,则在形成液晶单元的情况下,液晶取向膜中的倍半硅氧烷流入液晶中,显示特性容易下降。将残膜率为90%以上的情况评价为“良好(○)”,将80%以上且小于90%的情况评价为“可(△)”,将小于80%的情况评价为“不良(×)”。其结果为,该实施例中为残膜率“良好(○)”。
(4)液晶单元的制造
制作图2所示的FFS型液晶显示元件10。首先,将在其中一面具有电极对的玻璃基板11a、与未设置电极的对向玻璃基板11b作为一对,所述电极对依次形成有不具有图案的底电极15、作为绝缘层14的氮化硅膜、以及经图案化为梳齿状的顶电极13,在玻璃基板11a的具有透明电极的面、与对向玻璃基板11b的其中一面,分别使用旋转器来涂布所述(1)中制备的液晶取向剂(A-1),形成涂膜。继而,将该涂膜在80℃的加热板进行1分钟预烘烤后,在将库内进行了氮气置换的烘箱中以230℃进行加热(后烘烤)15分钟,形成平均膜厚为0.1μm的涂膜。
将此处使用的顶电极13的平面示意图示于图3(a)及图3(b)中。此外,图3(a)为顶电极13的俯视图,图3(b)为图3(a)的虚线所包围的部分C1的放大图。本实施例中,使用具有电极的线宽d1为4μm、电极间的距离d2为6μm的顶电极的基板。另外,顶电极13是使用电极A、电极B、电极C及电极D的四系统的驱动电极。图4中示出所使用的驱动电极的构成。底电极15作为对四系统的驱动电极全部发挥作用的共通电极而工作,四系统的驱动电极的区域分别成为像素区域。
继而,对于这些涂膜的各表面,使用Hg-Xe灯以及格兰泰勒棱镜(Glan-Taylor prism),自基板法线方向照射300J/m2的包含313nm明线的偏光紫外线,获得包含液晶取向膜的一对基板。此时,偏光紫外线的照射方向设为自基板法线方向,以将偏光紫外线的偏光面投影至基板上的线段的方向成为图3(b)中的双箭头的方向的方式来设定偏光面方向,然后进行光照射处理。
接着,在所述基板中的其中一块基板的包含液晶取向膜的面的外周,通过网版印刷来涂布加入有直径为5.5μm的氧化铝球的环氧树脂粘接剂后,使一对基板的液晶取向膜面对向,以将偏光紫外线的偏光面投影至基板上的方向成为平行的方式重叠压接,在150℃下花1小时使粘接剂进行热硬化。继而,自液晶注入口向基板间隙中填充默克(Merck)公司制造的液晶“MLC-6221”后,利用环氧树脂粘接剂将液晶注入口密封。然后,为了去除液晶注入时的流动取向,将其加热至150℃后再缓缓冷却至室温。
然后,通过在基板的外侧两面贴合偏光板来制造FFS型液晶显示元件。此时,偏光板中的其中一块是以其偏光方向成为与液晶取向膜的偏光紫外线的偏光面在基板面上的射影方向平行的方式来贴附,另一块是以其偏光方向成为与刚才的偏光板的偏光方向正交的方式来贴附。
(5)电压保持率的评价
对于所述(4)中制造的FFS型液晶显示元件,在23℃下以0.5微秒的施加时间、2,000毫秒的跨度施加1V的电压后,测定自施加解除起2000毫秒后的电压保持率(VHR)。此外,使用东阳特克尼卡(Toyo Technica)(股)制造的VHR-1作为测定装置。将电压保持率为95%以上的情况评价为“良好(○)”,将90%以上且小于95%的情况评价为“可(△)”,将小于90%的情况评价为“不良(×)”。其结果为,该实施例中为电压保持率“良好(○)”的结果。
(6)液晶取向性的评价
对于所述制造的液晶显示元件,利用光学显微镜来观察施加/解除5V电压时的明暗变化中的异常区域的有无。将未观察到异常区域的情况评价为液晶取向性“良好(○)”,将观察到异常区域的情况评价为液晶取向性“不良(×)”。其结果为,该液晶显示元件中未观察到异常区域,液晶取向性为“良好(○)”。
[实施例2~实施例13以及比较例1、比较例2]
除了在实施例1中,将聚合体的种类及使用量分别设为如下述表5中所记载以外,以与实施例1相同的方式制备液晶取向剂,使用所述液晶取向剂来进行各种评价。将评价结果示于下述表5中。
[表5]
表5中,聚合体[P]的数值表示相对于液晶取向剂的制备中使用的倍半硅氧烷(倍半硅氧烷[L]或者作为其他聚合体的倍半硅氧烷)100质量份而言的各聚合体的调配比例(质量份)。
如表5所示,包含倍半硅氧烷[L]的实施例1~实施例13在印刷性、残膜率、电压保持率以及液晶取向性的评价中均为“良好”或“可”。与此相对,比较例1、比较例2较实施例而言更差。
- 还没有人留言评论。精彩留言会获得点赞!
- 液晶取向处理剂、液晶取向膜和液晶表示元件的制作方法与工艺
- 一种液晶取向剂、液晶取向膜以及液晶显示元件的制作方法与工艺
- 液晶取向剂、液晶取向膜和液晶表示元件的制作方法与工艺
- 展曲取向液晶透镜的制作方法与工艺
- 液晶取向剂、液晶取向膜、液晶元件、以及液晶取向膜及液晶元件的制造方法与流程
- 液晶取向剂、液晶取向膜和使用其的液晶显示元件的制造方法与工艺
- 横向电场驱动型液晶表示元件用液晶取向膜制造用组合物、使用了该组合物的液晶取向膜及其制造方法、以及具有液晶取向膜的液晶表示元件及其制造方法与流程
- 液晶取向剂、液晶取向膜和使用了其的液晶表示元件的制造方法与工艺
- 液晶取向剂、液晶取向膜和液晶表示元件的制造方法与工艺
- 液晶取向剂、液晶取向膜和液晶表示元件的制造方法与工艺