一种钨酸铋/氮化碳/磷酸铋复合光催化剂及其制备方法和应用与流程
本发明属于功能材料领域,涉及一种Bi2WO6/C3N4/BiPO4复合光催化剂及其制备方法和应用。
背景技术:
Bi2WO6的禁带宽度约为3eV,具有良好的可见光响应,是一种新型的可见光催化材料,可用于降解工业废水中的有机污染物。Bi2WO6在受到可见光照射时,电子能从价带跃迁至导带,在价带上留下空位,光生电子和空穴与O2和表面OH-结合,形成具有氧化性的超氧自由基·O2-和羟基自由基·OH等,超氧自由基和羟基自由基能分解水中的有机物分子,达到降解的效果。C3N4也是一种窄禁带的光催化材料,其禁带宽度约为2.7eV,对可见光具有良好的响应,并且具有片层状结构,有一定的吸附性能。BiPO4禁带宽度约为4eV,是一种宽禁带半导体,对紫外光有良好的响应,但对可见光几乎没有响应。Bi2WO6和C3N4的禁带宽度较小,电子-空穴容易发生复合,目前还没有将Bi2WO6、C3N4和BiPO4结合起来制备Bi2WO6/C3N4/BiPO4复合光催化剂的相关报道。
技术实现要素:
本发明的目的在于提供一种钨酸铋/氮化碳/磷酸铋复合光催化剂的制备方法,该方法能够制备出具有良好的光催化效果的Bi2WO6/C3N4/BiPO4复合光催化剂,可用于有机污染物降解和其他光催化等方面。
为了实现上述目的,本发明采用如下技术方案:
一种钨酸铋/氮化碳/磷酸铋复合光催化剂的制备方法,包括以下步骤:
步骤1:按摩尔比为1:2将Na2WO4和Bi(NO3)3溶于去离子水中,搅拌均匀,得到混合溶液A,混合溶液A依次经超声分散和磁力搅拌后移至水热反应釜中,进行微波水热反应,反应结束后将产物静置分层,对下层的沉淀物进行洗涤、干燥,得到Bi2WO6粉体;
步骤2:按质量比为1:(1~1.5)将三聚氰胺和尿素混合均匀后加入去离子水中,得到混合物,对混合物进行搅拌反应,反应结束后将产物烘干并研磨,再在马弗炉中煅烧,将煅烧产物磨细,得到C3N4粉体;
步骤3:按摩尔比为3:3:(4~5)将Na3PO4、Bi(NO3)3和硝酸溶于去离子水中,搅拌均匀,得到混合溶液B,混合溶液B依次经超声分散和磁力搅拌后移至水热反应釜中,进行微波水热反应,反应结束后将产物静置分层,对下层的沉淀物进行洗涤、干燥,得到BiPO4粉体;
步骤4:将步骤1制得的Bi2WO6粉体和步骤2制得的C3N4粉体按质量比为(1.2~4.5):(5~6)加入到无水甲醇中,超声分散后形成前驱溶液A,对前驱溶液A进行磁力搅拌,得到悬浊液A,将悬浊液A静置分层,对下层的沉淀物进行洗涤、干燥,得到Bi2WO6/C3N4复合粉体;
步骤5:将步骤3制得的BiPO4粉体和步骤4制得的Bi2WO6/C3N4复合粉体按质量比为(0.4~2.8):(9.6~7.2)加入到无水甲醇中,超声分散后形成前驱溶液B,对前驱溶液B进行磁力搅拌,得到悬浊液B,将悬浊液B静置分层,对下层的沉淀物进行洗涤、干燥,得到钨酸铋/氮化碳/磷酸铋复合光催化剂。
所述步骤1中混合溶液A中Bi元素的浓度为0.133~0.15mol/L;
所述步骤2中混合物中三聚氰胺和尿素的浓度为0.25~0.333g/mL;
所述步骤3中混合溶液B中Bi元素的浓度为0.067~0.084mol/L。
所述步骤1和步骤3中超声分散的时间为20~30min,超声功率为400~500W;
所述步骤4和步骤5中超声分散的时间为2~3h,超声功率为400~500W。
所述步骤1和步骤3中磁力搅拌的时间为2~3h,搅拌速度为200~300rad/min;
所述步骤2中搅拌反应的时间为3~4h,搅拌速度为200~300rad/min;
所述步骤4和步骤5中磁力搅拌的时间为20~24h,搅拌速度为200~300rad/min。
所述步骤1和步骤3中的微波水热反应是在300W的微波功率下,在180~200℃下微波水热反应0.5~1h;
所述步骤2中的煅烧温度为500~550℃,煅烧时间为3~4h。
所述步骤1、步骤3、步骤4和步骤5中的洗涤、干燥是将沉淀物分别用无水乙醇和去离子水清洗去除杂质,然后在70~80℃下干燥15~20h;
所述步骤2中的烘干温度为70~80℃,烘干时间为15~20h。
所述步骤4中前驱溶液A中Bi2WO6和C3N4的总浓度为0.1~0.15g/mL;
所述步骤5中前驱溶液B中BiPO4和Bi2WO6/C3N4的总浓度为0.1~0.15g/mL。
所述的钨酸铋/氮化碳/磷酸铋复合光催化剂的制备方法制得的钨酸铋/氮化碳/磷酸铋复合光催化剂,该复合光催化剂中钨酸铋的物相为正交相Bi2WO6,空间群为Pca21(29),磷酸铋的物相为单斜相BiPO4,空间群为P21/n(14),氮化碳为层状物;在300W氙灯的模拟太阳光照射条件下,其光催化活性是Bi2WO6粉体的1.75~2.17倍,是C3N4粉体的1.91~2.36倍。
在300W氙灯的模拟太阳光照射条件下,其对罗丹明B的降解速率为0.021~0.026min-1。
所述的钨酸铋/氮化碳/磷酸铋复合光催化剂在光催化降解有机污染物方面的应用。
相对于现有的技术,本发明具有以下有益效果:
1.本发明首次将超声搅拌法应用于Bi2WO6/C3N4/BiPO4复合光催化剂的制备。本发明提供的Bi2WO6/C3N4/BiPO4复合光催化剂制备方法,先用微波水热法制备出两种结晶程度良好的Bi2WO6粉体和BiPO4粉体,再将三聚氰胺和尿素共烧获得C3N4粉体,再用温和的超声搅拌法分两步制备Bi2WO6/C3N4/BiPO4复合光催化剂,工艺简单,复合过程在室温下进行,反应条件 温和,复合过程中Bi2WO6、C3N4和BiPO4三种粉体各自的物相保持不变,本发明所制备的复合粉体结晶度高,组分间形成异质结结构。
2.本发明所制备的Bi2WO6/C3N4/BiPO4复合光催化剂具有很好的光催化性能,可用于光催化降解有机污染物。本发明将Bi2WO6、C3N4和BiPO4这三种半导体相互复合,得到一种新型的光催化半导体,即Bi2WO6/C3N4/BiPO4复合光催化剂,该Bi2WO6/C3N4/BiPO4复合光催化剂既具有良好的吸附性能,并且在可见光和紫外光波段皆有光响应,同时,由于Bi2WO6、C3N4和BiPO4这三种半导体的价带导带位置不同,光生电子、空穴分离后能在导带、价带发生迁移,提高分离效率,进而提升Bi2WO6/C3N4/BiPO4复合光催化剂的光催化效率。
3.本发明所制备的Bi2WO6/C3N4/BiPO4复合光催化剂相对Bi2WO6和C3N4光催化剂的光催化性能有明显提升,在300W氙灯的模拟太阳光照射条件下,其光催化活性是Bi2WO6粉体的1.75~2.17倍,是C3N4粉体的1.91~2.36倍,在光催化降解有机污染物方面及其他光催化方面有广泛的应用前景。
附图说明
图1是本发明制备的Bi2WO6/C3N4/BiPO4复合光催化剂的XRD图;
图2是本发明制备的Bi2WO6/C3N4/BiPO4复合光催化剂的FT-IR图;
图3是本发明制备的Bi2WO6/C3N4/BiPO4复合光催化剂的SEM图,其中(a)为Bi2WO6粉体,(b)为BiPO4粉体,(c)为C3N4粉体,(d)为Bi2WO6/C3N4复合粉体,(e)、(f)为Bi2WO6/C3N4/BiPO4复合光催化剂。
图4是本发明制备的Bi2WO6/C3N4/BiPO4复合光催化剂的降解曲线图。
具体实施方式
下面结合附图和本发明较优的实施例对本发明做进一步详细说明。
实施例1
步骤1:将原料3mmol Na2WO4和6mmol Bi(NO3)3溶于去离子水中,剧烈搅拌20min,得到白色混合溶液,混合溶液中Bi元素的浓度为0.133mol/L,将混合溶液在超声仪中以400W的功率超声分散30min,再在常温下以300rad/min的转速磁力搅拌2h,最后转移至水热反应釜中,微波功率为300W,在180℃下微波水热反应1h,得到白色混合溶液,将混合溶液静置分层,去除上清液,将沉淀分别用无水乙醇和去离子水清洗3次,在80℃下干燥15h,得到白色Bi2WO6粉体;
步骤2:按质量比为1:1分别称取三聚氰胺和尿素,混合均匀后加入去离子水中,得到混合物,混合物中三聚氰胺和尿素的浓度为0.333g/mL,以250rad/min的转速搅拌混合物4h,然后在80℃的烘箱中烘15h,将干粉取出并充分研磨,最后在550℃的马弗炉中煅烧4h,得到黄色块体,将块体充分研磨,得到黄色C3N4粉体;
步骤3:将3mmol Na3PO4、3mmol Bi(NO3)3和5mL 1mol/L硝酸溶液溶于去离子水中,搅拌后得到白色混合溶液,混合溶液中Bi元素的浓度为0.067mol/L,将混合溶液在超声仪中以400W的功率超声分散30min,再在常温下以300rad/min的转速磁力搅拌2h,最后转移至水热反应釜中,微波功率为300W,在200℃下微波水热反应1h,得到白色悬浊液,将悬浊液静置分层,去除上清液,将沉淀分别用无水乙醇和去离子水清洗3次,在80℃下干燥15h,得到白色BiPO4粉体;
步骤4:将步骤1中所得Bi2WO6粉体和步骤2中所得C3N4粉体以3.6:6的质量比加入到无水甲醇中,以400W的功率超声分散3h,形成前驱溶液,前驱溶液中Bi2WO6和C3N4的总浓度为0.15g/mL,以300rad/min的转速对前驱溶液磁力搅拌24h,得到黄色悬浊液,将悬浊液静置分层,去除上清液,所得黄色沉淀用无水乙醇和去离子水各洗涤三次,在80℃下干燥15h, 得到Bi2WO6/C3N4复合粉体。
步骤5:将步骤3中微波水热合成的BiPO4粉体和步骤4中合成的Bi2WO6/C3N4复合粉体按0.4:9.6的质量比加入到无水甲醇中,以400W的功率超声分散3h,形成前驱溶液,前驱溶液中Bi2WO6/C3N4和BiPO4的总浓度为0.15g/mL,以300rad/min的转速对前驱溶液磁力搅拌24h,得到黄色悬浊液,将悬浊液静置一段时间,去除上清液,所得黄色沉淀用无水乙醇和去离子水各洗涤三次,在80℃下干燥15h,得到Bi2WO6/C3N4/BiPO4复合光催化剂。
实施例2
步骤1:将原料3mmol Na2WO4和6mmol Bi(NO3)3溶于去离子水中,剧烈搅拌20min,得到白色混合溶液,混合溶液中Bi元素的浓度为0.14mol/L,将混合溶液在超声仪中以500W的功率超声分散20min,再在常温下以200rad/min的转速磁力搅拌3h,最后转移至水热反应釜中,微波功率为300W,在200℃下微波水热反应0.5h,得到白色混合溶液,将混合溶液静置分层,去除上清液,将沉淀分别用无水乙醇和去离子水清洗3次,在70℃下干燥16h,得到白色Bi2WO6粉体;
步骤2:按质量比为1:1.1分别称取三聚氰胺和尿素,混合均匀后加入去离子水中,得到混合物,混合物中三聚氰胺和尿素的浓度为0.25g/mL,以200rad/min的转速搅拌混合物3.5h,然后在70℃的烘箱中烘16h,将干粉取出并充分研磨,最后在500℃的马弗炉中煅烧3.8h,得到黄色块体,将块体充分研磨,得到黄色C3N4粉体;
步骤3:将3mmol Na3PO4、3mmol Bi(NO3)3和4mL 1mol/L硝酸溶液溶于去离子水中,搅拌后得到白色混合溶液,混合溶液中Bi元素的浓度为0.07mol/L,将混合溶液在超声仪中以500W的功率超声分散20min,再在常温下以200rad/min的转速磁力搅拌3h,最后转移至水热反应釜中,微波功率为300W,在190℃下微波水热反应0.6h,得到白色悬浊液,将悬浊液静置分层,去除上清液,将沉淀分别用无水乙醇和去离子水清洗3次,在70℃下干燥16h,得到 白色BiPO4粉体;
步骤4:将步骤1中所得Bi2WO6粉体和步骤2中所得C3N4粉体以2.8:6的质量比加入到无水甲醇中,以500W的功率超声分散2h,形成前驱溶液,前驱溶液中Bi2WO6和C3N4的总浓度为0.1g/mL,以200rad/min的转速对前驱溶液磁力搅拌23h,得到黄色悬浊液,将悬浊液静置分层,去除上清液,所得黄色沉淀用无水乙醇和去离子水各洗涤三次,在70℃下干燥16h,得到Bi2WO6/C3N4复合粉体。
步骤5:将步骤3中微波水热合成的BiPO4粉体和步骤4中合成的Bi2WO6/C3N4复合粉体按1.2:8.8的质量比加入到无水甲醇中,以500W的功率超声分散2h,形成前驱溶液,前驱溶液中Bi2WO6/C3N4和BiPO4的总浓度为0.1g/mL,以200rad/min的转速对前驱溶液磁力搅拌23h,得到黄色悬浊液,将悬浊液静置一段时间,去除上清液,所得黄色沉淀用无水乙醇和去离子水各洗涤三次,在70℃下干燥16h,得到Bi2WO6/C3N4/BiPO4复合光催化剂。
实施例3
步骤1:将原料3mmol Na2WO4和6mmol Bi(NO3)3溶于去离子水中,剧烈搅拌20min,得到白色混合溶液,混合溶液中Bi元素的浓度为0.145mol/L,将混合溶液在超声仪中以450W的功率超声分散25min,再在常温下以250rad/min的转速磁力搅拌2.5h,最后转移至水热反应釜中,微波功率为300W,在190℃下微波水热反应0.6h,得到白色混合溶液,将混合溶液静置分层,去除上清液,将沉淀分别用无水乙醇和去离子水清洗3次,在75℃下干燥17h,得到白色Bi2WO6粉体;
步骤2:按质量比为1:1.2分别称取三聚氰胺和尿素,混合均匀后加入去离子水中,得到混合物,混合物中三聚氰胺和尿素的浓度为0.28g/mL,以300rad/min的转速搅拌混合物3h,然后在75℃的烘箱中烘17h,将干粉取出并充分研磨,最后在510℃的马弗炉中煅烧3.6h,得到黄色块体,将块体充分研磨,得到黄色C3N4粉体;
步骤3:将3mmol Na3PO4、3mmol Bi(NO3)3和4.5mL 1mol/L硝酸溶液溶于去离子水中,搅拌后得到白色混合溶液,混合溶液中Bi元素的浓度为0.075mol/L,将混合溶液在超声仪中以450W的功率超声分散25min,再在常温下以250rad/min的转速磁力搅拌2.5h,最后转移至水热反应釜中,微波功率为300W,在180℃下微波水热反应0.7h,得到白色悬浊液,将悬浊液静置分层,去除上清液,将沉淀分别用无水乙醇和去离子水清洗3次,在75℃下干燥17h,得到白色BiPO4粉体;
步骤4:将步骤1中所得Bi2WO6粉体和步骤2中所得C3N4粉体以2:6的质量比加入到无水甲醇中,以450W的功率超声分散2.5h,形成前驱溶液,前驱溶液中Bi2WO6和C3N4的总浓度为0.11g/mL,以250rad/min的转速对前驱溶液磁力搅拌22h,得到黄色悬浊液,将悬浊液静置分层,去除上清液,所得黄色沉淀用无水乙醇和去离子水各洗涤三次,在75℃下干燥17h,得到Bi2WO6/C3N4复合粉体。
步骤5:将步骤3中微波水热合成的BiPO4粉体和步骤4中合成的Bi2WO6/C3N4复合粉体按2:8的质量比加入到无水甲醇中,以450W的功率超声分散2.5h,形成前驱溶液,前驱溶液中Bi2WO6/C3N4和BiPO4的总浓度为0.11g/mL,以250rad/min的转速对前驱溶液磁力搅拌22h,得到黄色悬浊液,将悬浊液静置一段时间,去除上清液,所得黄色沉淀用无水乙醇和去离子水各洗涤三次,在75℃下干燥17h,得到Bi2WO6/C3N4/BiPO4复合光催化剂。
实施例4
步骤1:将原料3mmol Na2WO4和6mmol Bi(NO3)3溶于去离子水中,剧烈搅拌20min,得到白色混合溶液,混合溶液中Bi元素的浓度为0.15mol/L,将混合溶液在超声仪中以420W的功率超声分散28min,再在常温下以220rad/min的转速磁力搅拌2.8h,最后转移至水热反应釜中,微波功率为300W,在195℃下微波水热反应0.7h,得到白色混合溶液,将混合溶液静置分层,去除上清液,将沉淀分别用无水乙醇和去离子水清洗3次,在72℃下干燥18h,得到白 色Bi2WO6粉体;
步骤2:按质量比为1:1.3分别称取三聚氰胺和尿素,混合均匀后加入去离子水中,得到混合物,混合物中三聚氰胺和尿素的浓度为0.30g/mL,以220rad/min的转速搅拌混合物3.2h,然后在72℃的烘箱中烘18h,将干粉取出并充分研磨,最后在520℃的马弗炉中煅烧3.2h,得到黄色块体,将块体充分研磨,得到黄色C3N4粉体;
步骤3:将3mmol Na3PO4、3mmol Bi(NO3)3和4.2mL 1mol/L硝酸溶液溶于去离子水中,搅拌后得到白色混合溶液,混合溶液中Bi元素的浓度为0.08mol/L,将混合溶液在超声仪中以420W的功率超声分散28min,再在常温下以220rad/min的转速磁力搅拌2.8h,最后转移至水热反应釜中,微波功率为300W,在185℃下微波水热反应0.8h,得到白色悬浊液,将悬浊液静置分层,去除上清液,将沉淀分别用无水乙醇和去离子水清洗3次,在72℃下干燥18h,得到白色BiPO4粉体;
步骤4:将步骤1中所得Bi2WO6粉体和步骤2中所得C3N4粉体以1.2:6的质量比加入到无水甲醇中,以420W的功率超声分散2.8h,形成前驱溶液,前驱溶液中Bi2WO6和C3N4的总浓度为0.12g/mL,以220rad/min的转速对前驱溶液磁力搅拌24h,得到黄色悬浊液,将悬浊液静置分层,去除上清液,所得黄色沉淀用无水乙醇和去离子水各洗涤三次,在72℃下干燥18h,得到Bi2WO6/C3N4复合粉体。
步骤5:将步骤3中微波水热合成的BiPO4粉体和步骤4中合成的Bi2WO6/C3N4复合粉体按2.8:7.2的质量比加入到无水甲醇中,以420W的功率超声分散2.8h,形成前驱溶液,前驱溶液中Bi2WO6/C3N4和BiPO4的总浓度为0.12g/mL,以220rad/min的转速对前驱溶液磁力搅拌22.5h,得到黄色悬浊液,将悬浊液静置一段时间,去除上清液,所得黄色沉淀用无水乙醇和去离子水各洗涤三次,在72℃下干燥18h,得到Bi2WO6/C3N4/BiPO4复合光催化剂。
实施例5
步骤1:将原料3mmol Na2WO4和6mmol Bi(NO3)3溶于去离子水中,剧烈搅拌20min,得到白色混合溶液,混合溶液中Bi元素的浓度为0.137mol/L,将混合溶液在超声仪中以480W的功率超声分散22min,再在常温下以280rad/min的转速磁力搅拌2.2h,最后转移至水热反应釜中,微波功率为300W,在185℃下微波水热反应0.8h,得到白色混合溶液,将混合溶液静置分层,去除上清液,将沉淀分别用无水乙醇和去离子水清洗3次,在78℃下干燥19h,得到白色Bi2WO6粉体;
步骤2:按质量比为1:1.4分别称取三聚氰胺和尿素,混合均匀后加入去离子水中,得到混合物,混合物中三聚氰胺和尿素的浓度为0.31g/mL,以280rad/min的转速搅拌混合物3.8h,然后在78℃的烘箱中烘19h,将干粉取出并充分研磨,最后在530℃的马弗炉中煅烧3.5h,得到黄色块体,将块体充分研磨,得到黄色C3N4粉体;
步骤3:将3mmol Na3PO4、3mmol Bi(NO3)3和4.8mL 1mol/L硝酸溶液溶于去离子水中,搅拌后得到白色混合溶液,混合溶液中Bi元素的浓度为0.078mol/L,将混合溶液在超声仪中以480W的功率超声分散22min,再在常温下以280rad/min的转速磁力搅拌2.2h,最后转移至水热反应釜中,微波功率为300W,在195℃下微波水热反应0.9h,得到白色悬浊液,将悬浊液静置分层,去除上清液,将沉淀分别用无水乙醇和去离子水清洗3次,在78℃下干燥19h,得到白色BiPO4粉体;
步骤4:将步骤1中所得Bi2WO6粉体和步骤2中所得C3N4粉体以4.5:5的质量比加入到无水甲醇中,以480W的功率超声分散2.2h,形成前驱溶液,前驱溶液中Bi2WO6和C3N4的总浓度为0.13g/mL,以280rad/min的转速对前驱溶液磁力搅拌20h,得到黄色悬浊液,将悬浊液静置分层,去除上清液,所得黄色沉淀用无水乙醇和去离子水各洗涤三次,在78℃下干燥19h,得到Bi2WO6/C3N4复合粉体。
步骤5:将步骤3中微波水热合成的BiPO4粉体和步骤4中合成的Bi2WO6/C3N4复合粉体 按0.5:9.5的质量比加入到无水甲醇中,以480W的功率超声分散2.2h,形成前驱溶液,前驱溶液中Bi2WO6/C3N4和BiPO4的总浓度为0.13g/mL,以280rad/min的转速对前驱溶液磁力搅拌20h,得到黄色悬浊液,将悬浊液静置一段时间,去除上清液,所得黄色沉淀用无水乙醇和去离子水各洗涤三次,在78℃下干燥19h,得到Bi2WO6/C3N4/BiPO4复合光催化剂。
实施例6
步骤1:将原料3mmol Na2WO4和6mmol Bi(NO3)3溶于去离子水中,剧烈搅拌20min,得到白色混合溶液,混合溶液中Bi元素的浓度为0.135mol/L,将混合溶液在超声仪中以460W的功率超声分散26min,再在常温下以260rad/min的转速磁力搅拌2.4h,最后转移至水热反应釜中,微波功率为300W,在180℃下微波水热反应0.9h,得到白色混合溶液,将混合溶液静置分层,去除上清液,将沉淀分别用无水乙醇和去离子水清洗3次,在80℃下干燥20h,得到白色Bi2WO6粉体;
步骤2:按质量比为1:1.5分别称取三聚氰胺和尿素,混合均匀后加入去离子水中,得到混合物,混合物中三聚氰胺和尿素的浓度为0.32g/mL,以260rad/min的转速搅拌混合物3.6h,然后在80℃的烘箱中烘20h,将干粉取出并充分研磨,最后在540℃的马弗炉中煅烧3h,得到黄色块体,将块体充分研磨,得到黄色C3N4粉体;
步骤3:将3mmol Na3PO4、3mmol Bi(NO3)3和4.6mL 1mol/L硝酸溶液溶于去离子水中,搅拌后得到白色混合溶液,混合溶液中Bi元素的浓度为0.084mol/L,将混合溶液在超声仪中以460W的功率超声分散26min,再在常温下以260rad/min的转速磁力搅拌2.4h,最后转移至水热反应釜中,微波功率为300W,在200℃下微波水热反应0.5h,得到白色悬浊液,将悬浊液静置分层,去除上清液,将沉淀分别用无水乙醇和去离子水清洗3次,在80℃下干燥20h,得到白色BiPO4粉体;
步骤4:将步骤1中所得Bi2WO6粉体和步骤2中所得C3N4粉体以4:5的质量比加入到无 水甲醇中,以460W的功率超声分散2.4h,形成前驱溶液,前驱溶液中Bi2WO6和C3N4的总浓度为0.14g/mL,以260rad/min的转速对前驱溶液磁力搅拌21h,得到黄色悬浊液,将悬浊液静置分层,去除上清液,所得黄色沉淀用无水乙醇和去离子水各洗涤三次,在80℃下干燥20h,得到Bi2WO6/C3N4复合粉体。
步骤5:将步骤3中微波水热合成的BiPO4粉体和步骤4中合成的Bi2WO6/C3N4复合粉体按1:9的质量比加入到无水甲醇中,以460W的功率超声分散2.4h,形成前驱溶液,前驱溶液中Bi2WO6/C3N4和BiPO4的总浓度为0.14g/mL,以260rad/min的转速对前驱溶液磁力搅拌21h,得到黄色悬浊液,将悬浊液静置一段时间,去除上清液,所得黄色沉淀用无水乙醇和去离子水各洗涤三次,在80℃下干燥20h,得到Bi2WO6/C3N4/BiPO4复合光催化剂。
采用XRD测定Bi2WO6/C3N4/BiPO4复合光催化剂制备方法的物相组成结构;用傅里叶红外光谱表征光催化剂的官能团种类和所处的化学环境进行表征;表面形貌由S-4800型场发射扫描电子显微镜进行表征;在XPA-7型光催化反应仪中进行罗丹明B的降解实验,表征光催化剂的光催化性能。
图1为本发明制备的Bi2WO6/C3N4/BiPO4复合光催化剂的XRD图,从下到上依次为Bi2WO6、C3N4和BiPO4的质量比为2.8:6:1.2、2:6:2和1.2:6:2.8的Bi2WO6/C3N4/BiPO4复合光催化剂。其中Bi2WO6/C3N4/BiPO4复合光催化剂在2θ=28.34°,32.94°,47.18°,55.96°左右出现的衍射峰分别对应的是正交相Bi2WO6(PDF No.79-2381)的(131)、(002)、(202)、(133)晶面,在2θ=19.06°,21.40°,27.20°,29.14°,31.24°处的衍射峰分别对应的是单斜相BiPO4(PDFNO.15-0767)的(011)、(-111)、(200)、(120)、(012)晶面。因C3N4为片状材料,在XRD图中无法测到。说明本发明成功合成了Bi2WO6/C3N4/BiPO4复合光催化剂。
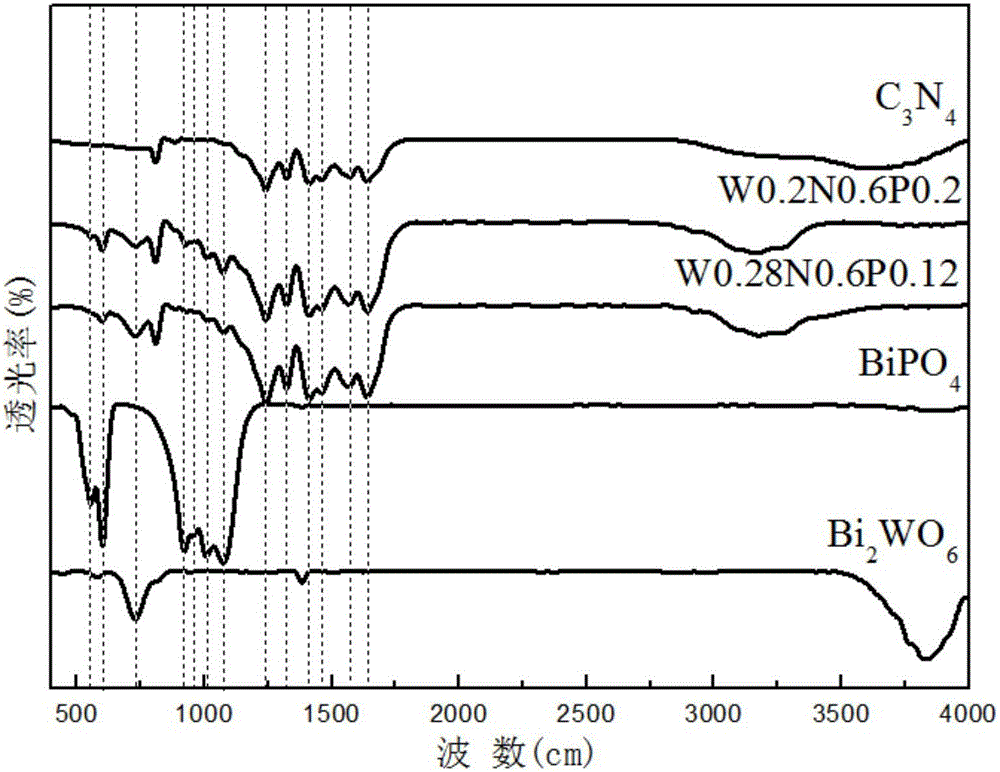
图2是Bi2WO6/C3N4/BiPO4复合光催化剂的FT-IR图,在730cm-1处对应的是Bi2WO6的W-O伸缩震动吸收峰,1073cm-1到921cm-1对应的是(PO4)的伸缩振动吸收峰,600cm-1到554cm-1对应的是(PO4)的弯曲振动吸收峰,1414~1640cm-1处的吸收峰对应的是碳氮杂环中C-N和C=N的伸缩振动峰,3500~4000cm-1处的吸收峰属于g-C3N4合成过程中未完全分解的N-H和表面吸附的H2O的吸收峰,进一步说明形成Bi2WO6/C3N4/BiPO4复合光催化剂。
图3为Bi2WO6/C3N4/BiPO4复合光催化剂的SEM图,Bi2WO6呈花状,花瓣边缘呈锯齿状,颗粒尺寸为2μm(图3a),BiPO4的形貌是表面光滑方形棒状颗粒,尺寸在2~0.2μm左右,直径在0.4~0.15μm(图3b),纯相C3N4为多孔块体(图3c),复合后光催化剂的C3N4尺寸减小,附着在Bi2WO6表面(图3d),BiPO4颗粒嵌插在Bi2WO6/C3N4上(图3e、f)。
图4是本发明制备的Bi2WO6/C3N4/BiPO4复合光催化剂的降解曲线图,在模拟太阳光照射下(300W氙灯),Bi2WO6、C3N4和BiPO4的质量比为3.6:6:0.4、2.8:6:1.2、2:6:2和1.2:6:2.8的Bi2WO6/C3N4/BiPO4复合光催化剂对RhB的降解速率k依次为0.023cm-1、0.026cm-1、0.025cm-1和0.021cm-1,Bi2WO6粉体对粉体RhB的降解速率k为0.012cm-1,C3N4粉体对RhB的降解速率k为0.011cm-1,Bi2WO6/C3N4/BiPO4复合光催化剂光催化活性是Bi2WO6降解速率的1.75~2.17倍,是C3N4反应速率的1.91~2.36倍,Bi2WO6/C3N4/BiPO4复合光催化剂的光催化性能有了明显提升。
以上所述内容是结合具体的优选实施方式对本发明所作的进一步详细说明,不是全部或唯一的实施方式,本领域普通技术人员通过阅读本发明说明书而对本发明技术方案采取的任何等效的变换,均为本发明的权利要求所涵盖。
- 还没有人留言评论。精彩留言会获得点赞!