砷钒氧簇基多酸离子液体及其制备方法和应用与流程
本发明属于离子液体技术领域,特别是涉及一种砷钒氧簇基多酸离子液体及其制备方法和应用。
背景技术:
探索多金属氧簇的合成方法,或者对已有方法进行优化,得到新颖的结构或者性能得到提高的化合物,是科学家不断追求的目标。19世纪初,离子液体以其独特的溶解性、操作安全性、环境友好性等特点进入科学家们的视野,在新型多酸的合成中离子液体既可以作为溶剂,也可以作为产物的构成组分,起到合成模板剂等的作用,这些特点无疑为多酸化学家合成新型多酸化合物提供了更多的机遇。
由于离子液体可以改变反应途径,从而能更好的发挥催化活性,作为反应媒介在Heck反应、催化脱硫等方面都有很好的表现。而多酸作为兼具酸性和氧化性特质的优秀催化剂早在1972年就被应用。多酸型离子液体作为二者优良的结合体,目前在催化领域的研究还处于初期阶段,该体系具有很大的发展空间。
2009年以来,王军等人以Keggin型的多钨酸离子液体进行了催化酯化反应、催化脱硫反应等研究,发现该类体系具有很好的催化效果。据文献调研发现,目前针对砷钒氧簇基多酸离子液体的研究还处于空白。
技术实现要素:
为此,本发明提供了一种砷钒氧簇基多酸离子液体,所述多酸离子液体的化学式如下式(1)所示:
[CnMIm]4[HAs8V12O40(Cl)] (1)
其中:n为2、3、5、6或7
优选地,所述多酸离子液体的化学式为[C2MIm]4[HAs8V12O40(Cl)]、[C3MIm]4[HAs8V12O40(Cl)]、[C5MIm]4[HAs8V12O40(Cl)]、[C6MIm]4[HAs8V12O40(Cl)]或[C7MIm]4[HAs8V12O40(Cl)]。
优选地,所述多酸离子液体的阴离子具有以氯离子为模板剂的As8V12O40砷钒氧簇笼状结构。
优选地,本发明还提供了该砷钒氧簇基多酸离子液体的制备方法,具体步骤如下:
分别称取偏钒酸钠、亚砷酸钠、盐酸联氨和[CnMIm]Br,n的定义同式(1),所述偏钒酸钠、亚砷酸钠、盐酸联氨、[CnMIm]Br的摩尔比为:1∶1∶2∶2;
将偏钒酸钠、亚砷酸钠分别加水溶解,形成偏钒酸钠溶液和亚砷酸钠溶液,将上述偏钒酸钠溶液和亚砷酸钠溶液混合搅拌30min后,加入盐酸联氨,搅拌30min,沉淀过滤,得到清液,在清液中加入[CnMIm]Br,室温条件下反应3小时,过滤得到沉淀,洗涤,得到所述砷钒氧簇基多酸离子液体。
更优选地,所述偏钒酸钠溶液的浓度为0.2~0.5mol/L,所述亚砷酸钠溶液的浓度为0.2~0.5mol/L。
本发明提供的砷钒氧簇基多酸离子液体,可用作光催化剂的应用。
本发明提供的砷钒氧簇多酸离子液体制备方法简单,所制得的多酸离子液体,溶解性好,具有很高的光催化活性和选择性。该方法首次将砷钒氧簇笼状结构引入到多酸离子液体的结构中,所制得的多酸离子液体在包括可见光在内的较宽范围内都具有良好吸光性能,在可见光的照射下即可实现有机染料的光催化降解。
附图说明
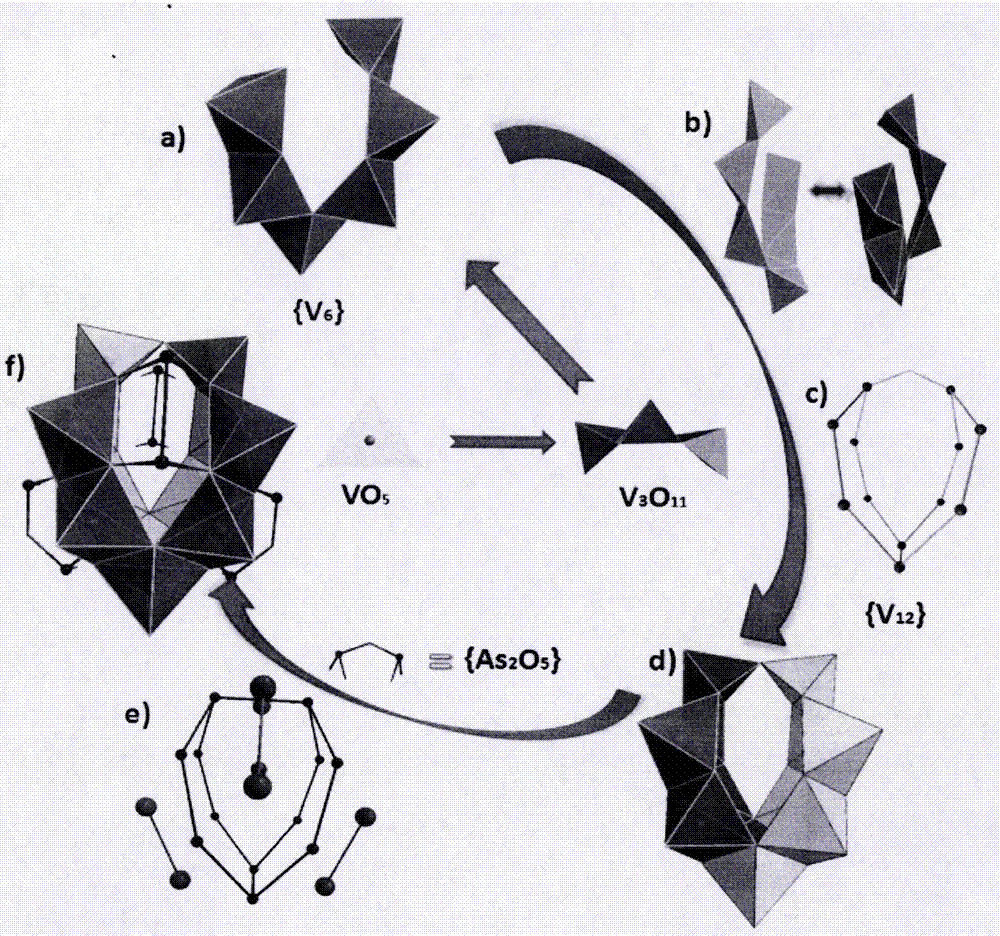
图1为本发明实施例中多酸离子液体中阴离子的笼状结构图;
图2为本发明实施例中多酸离子液体中阴离子的笼状结构的组装过程图;
图3为本发明实施例提供的化合物1-5的红外光谱图;
图4为本发明实施例1提供的化合物1的电子吸收光谱图;
图5为本发明实施例2提供的化合物2的电子吸收光谱图;
图6为本发明实施例3提供的化合物3的电子吸收光谱图;
图7为本发明实施例4提供的化合物4的电子吸收光谱图;
图8为本发明实施例5提供的化合物5的电子吸收光谱图;
图9为本发明实施例1提供的化合物1的光学禁带宽度图;
图10为本发明实施例2提供的化合物2的光学禁带宽度图;
图11为本发明实施例3提供的化合物3的光学禁带宽度图;
图12为本发明实施例4提供的化合物4的光学禁带宽度图;
图13为本发明实施例5提供的化合物5的光学禁带宽度图;
图14为本发明实施例1提供的化合物1的光催化降解RhB的紫外吸收随时间变化曲线图;
图15为本发明实施例2提供的化合物2的光催化降解RhB的紫外吸收随时间变化曲线图;
图16为本发明实施例3提供的化合物3的光催化降解RhB的紫外吸收随时间变化曲线图;
图17为本发明实施例4提供的化合物4的光催化降解RhB的紫外吸收随时间变化曲线图;
图18为本发明实施例5提供的化合物5的光催化降解RhB的紫外吸收随时间变化曲线图。
具体实施方式
为了使本领域技术人员更好地理解本发明的技术方案能予以实施,下面结合具体实施例对本发明作进一步说明,但所举实施例不作为对本发明的限定。
需要说明的是,以下实施例中所用到的试剂若无特别说明,均为常规试剂,可在市场上购买得到,且以下实施例中所有使用的化学试剂均为分析纯,使用前没有经过进一步的纯化;所涉及的相关化合物的制备方法或检测方法,若无特别说明,均为常规方法。
以下实施例中所涉及的测试,其中:C、H、N含量采用Perkin-Elmer 240C型元素分析仪测定;红外光谱采用Nicolet170傅立叶红外光谱仪测定(记录范围4000~400cm-1、KBr压片);晶体结构采用Bruker APEX II CCD型单晶X-射线衍射仪测定;粉末XRD用荷兰飞利浦X-PertPro粉末X-射线衍射仪测定;TG-DTA采用Perkin Elmer-7差热热重分析仪测定(由室温开始升温到1000℃,升温速度10℃/min,氮气气氛);紫外-可见光谱采用HITACHI U-4100分光光度计测定,液体紫外记录范围为800~200nm,固体紫外测定范围为2500~200nm;光催化降解有机染料采用南京胥江机电厂XPA型光催化反应仪(500W汞灯,420nm滤光片)测定。
实施例1
[C2MIm]4[HAs8V12O40(Cl)]的合成
分别称取0.63g偏钒酸钠、0.65g亚砷酸钠、1.049g盐酸联氨和1.911g[C2MIm]Br,将偏钒酸钠、亚砷酸钠分别溶解在10ml蒸馏水中,形成0.5mol/L的偏钒酸钠溶液和0.5mol/L的亚砷酸钠溶液,将上述偏钒酸钠溶液和亚砷酸钠溶液混合后搅拌30min,加入盐酸联氨,搅拌30min,沉淀过滤,得到清液,在清液中加入[C2MIm]Br,室温条件下反应3小时,过滤,蒸馏水洗涤3次,得到粉末状目标产物,产率为56%。
经元素分析仪测试,该化合物化学式为C24H45N8O40As8V12Cl,元素分析结果为:理论值C,12.36;H,1.94;N,4.81;测量值C,11.79;H,2.04;N,4.59。
实施例2
[C3MIm]4[HAs8V12O40(Cl)]的合成
该化合物的具体合成方法和实施例1相同,不同之处仅在于,将[C2MIm]Br替换为[C3MIm]Br,且[C3MIm]Br的反应摩尔比与[C2MIm]Br相同,得到的粉末状目标产物,产率为53%。
经元素分析仪测试,该化合物化学式为C28H53N8O40As8V12Cl,元素分析结果为:理论值:C,14.08;H,2.24;N,4.69;测量值:C,13.90;H,2.41;N,4.76。
实施例3
[C5MIm]4[HAs8V12O40(Cl)]的合成
该化合物的具体合成方法和实施例1相同,不同之处仅在于,将[C2MIm]Br替换为[C5MIm]Br,且[C5MIm]Br的反应摩尔比与[C2MIm]Br相同,得到的粉末状目标产物,产率为60%。
经元素分析仪测试,该化合物化学式为C36H69N8O40As8V12Cl,元素分析(%)结果为:理论值:C,17.30;H,2.78;N,4.48;测量值C,17.07;H,2.93;N,4.68。
实施例4
[C6MIm]4[HAs8V12O40(Cl)]的合成
该化合物的具体合成方法和实施例1相同,不同之处仅在于,将[C2MIm]Br替换为[C6MIm]Br,且[C6MIm]Br的反应摩尔比与[C2MIm]Br相同,得到的粉末状目标产物,产率为55%。
经元素分析仪测试,该化合物化学式为C40H77N8O40As8V12Cl,元素分析结果为:理论值:C,18.80;H,3.04;N,4.38;测量值:C,18.47;H,3.20;N,4.51。
实施例5
[C7MIm]4[HAs8V12O40(Cl)]的合成
该化合物的具体合成方法和实施例1相同,不同之处仅在于,将[C2MIm]Br替换为[C7MIm]Br,且[C7MIm]Br的反应摩尔比与[C2MIm]Br相同,得到的粉末状目标产物,产率为43%。
经元素分析仪测试,该化合物化学式为C44H85N8O40As8V12Cl,元素分析结果为:理论值:C,20.23;H,3.28;N,4.29;测量值:C,19.62;H,3.42;N,4.44。
具体的实施例1-5所制得的化合物分别称为化合物1-5,为了获取化合物1-5的结构信息,制备了化合物[(Butyl)4N]4[As8V12O40(N2H4)]的单晶样品,并进行了结构测定。具体内容如下:
[(Butyl)4N]3[H2As8V12O40(Cl)]的合成
分别称取0.63g偏钒酸钠、0.65g亚砷酸钠、1.049g盐酸联氨和3.224g四丁基溴化铵,将偏钒酸钠、亚砷酸钠分别溶解在10ml蒸馏水中,形成0.5mol/L的偏钒酸钠溶液和0.5mol/L的亚砷酸钠溶液,将上述偏钒酸钠溶液和亚砷酸钠溶液混合后搅拌30min,加入盐酸联氨,搅拌30min,沉淀过滤,得到清液,在清液中加入四丁基溴化铵,室温条件下反应3小时,过滤得到墨绿色沉淀,蒸馏水洗涤3次,DMF溶解重结晶,3周以后得到墨绿色块状晶体化合物,即为目标产物,经计算产率为56%。
经元素分析仪和IR测试,该化合物化学式为C48H110N3O40As8V12Cl,元素分析(%),理论值:C,22.04;H,4.24;N,1.61;测量值:C,21.85;H,4.06;N,1.48;IR(cm-1,KBr pellet):3440(s),2924(m),2349(w),2055(w),2015(m),1631(m),1402(m),1091(w),967(s),699(s),664(m),384(m)。
对获得的样品进行晶体结构测定,具体选择尺寸合适的单晶,以的Mo-Kα射线和ω-2θ扫描方式,在Bruker APEX II CCD X-射线单晶衍射仪(带有石墨单色器)上进行衍射实验。衍射强度通过Lp因子和经验吸收进行校正;晶体结构解析使用SHELXTL-97软件处理;晶体结构通过直接法解析。最后一轮修正(对所有非氢原子坐标)使用全矩阵最小二乘法各向异性修正和次级消光处理。用直接法得到所有非氢原子坐标,所有氢原子均为理论加氢并采用各向同性热参数和跨式模型进行修正。
化合物[(Butyl)4N]3[H2As8V12O40(Cl)]的晶体学参数为:单斜晶系,空间群P21/c,晶胞参数:β=96.525(4)°,Z=2,Dc=1.724g/cm3,μ=3.745mm-1,F(000)=2590,最大和最小残留电子密度峰分别为
具体的,晶体结构解析表明,[(Butyl)4N]3[H2As8V12O40(Cl)]结晶属于单斜晶系,最小不对称单元由1个[H2AS8V12O40(Cl)]3-阴离子和3个四丁基铵阳离子组成,Cl-为模板剂填充在聚阴离子笼中,阴离子的笼状结构具体如图1所示。
砷钒氧簇阴离子是由12个{VO5}基团和8个{AsO3}基团构成。在该化合物中12个{VO5}单元通过共边形成4个{V3O11}单元,2个{V3O11}单元通过共边形成1个{V6O20}单元,具体如图2所示,2个{V6O20}单元通过共边和共顶点构成{V12O30}单元,具体如图2中的c和d,该结构中的钒均采用五配位的{VO5}四方锥构型,端氧居于锥顶,V-Ot键长范围为μ2-O和μ3-O桥氧位于锥底,其V-Ob键长范围为该结构中的8个{AsO3}单元通过共顶点构成4个{As2O5}单元,每个{As2O5}单元通过4个氧原子分别扣在4个{V6O20}单元面上,构成[As8V12O40]4-阴离子,具体如图2中的f。结构中的砷原子均采用与3个氧原子配位,As-O键长范围为经价键计算,化合物中的钒均显示+5价,砷显示+3价,因此整个阴离子骨架为-4价。
对实施例1-5所制得的化合物进行红外测试,得到5个化合物的红外光谱图,具体如图3所示,
和化合物[(Butyl)4N]3[H2As8V12O40(Cl)]相似,化合物1-5在999、876、834、694、609cm-1处具有红外吸收特征峰,相对强度也基本一致,999cm-1处归属为V-Ot的红外吸收峰,876、834、694cm-1处的吸收峰可归属为M-O-M的红外吸收峰(M=V、As)。由此推断化合物1-5与化合物[(Butyl)4N]3[H2As8V12O40(Cl)]阴离子结构相同。
化合物1-5均在3100-3000cm-1有红外吸收峰,归属为芳环的C-H伸缩振动峰,同时化合物1-5分别在1600-1400cm-1间有振动峰,可归属为芳环的不饱和C=C键的伸缩振动,因此推断化合物1-5中存在[CnMIm]+(n=2、3、5、6、7)组分。
对5个化合物进行固体紫外-可见-近红外电子吸收光谱测定,5个化合物的电子吸收光谱图分别见图4-8所示,化合物1-5在紫外区至可见区的200-500nm范围内呈现出相似的吸收,最大吸收峰发生一定的偏移,这可能与化合物所处的环境有一定的影响,在可见-近红外的500-2000nm之间的吸收相同,最大吸收峰位置也基本不变。在紫外和可见区的吸收通常为O→V荷移跃迁,在可见-近红外区的吸收为V4+/V5+的价间荷移跃迁。
砷钒金属氧簇富含大量d电子,呈现出较好的光化学性能,因此研究了化合物1-5的光学禁带宽度,以固体紫外-可见光谱吸收参数为依据,经过转化得到Kubelka-Munk函数(F=(1-R)2/2R,R-特定波长下的漫反射率)和化合物的能带值,并将即为带隙宽度,计算化合物的带隙宽度是探究半导体潜质的基础途径。通过K-M函数与能带值作图,以其线性部分的延长线与能量轴的交点得到化合物1-5的带隙宽度分别为0.97、1.17、1.17、1.06、1.23ev,如图9-13所示,与常见的多金属氧簇的带隙宽度相比,该化合物的带隙宽度较窄,具有窄带半导体的潜质。
本发明提供的砷钒氧簇基多酸离子液体光催化降解罗丹明B的示例:
具体的试验方法为:配制1×10-4mol/LRhB溶液作为催化底物。分别称取化合物1-5各10mg加入到50mL配置好的底物溶液中,暗态搅拌40分钟,达到吸附脱附平衡后,取出一定量的悬浮液进行离心分离,测定RhB在553nm处的吸光度,记为I0。打开光源开始光照,随后在一定的时间间隔内取出一定量的反应体系进行离心分离,测定RhB的吸光度。RhB的降解率采用如下公式进行计算:K=[(I0-It)/I0]×100%(其中:K为照射一定时间后RhB的光降解率,I0为光照前溶液在553nm处的吸光度;It为光照时间为t时的吸光度)。
图14-18为化合物1-5作为催化剂,RhB的紫外吸收强度随时间的变化曲线,由图14-18可以看出,离子液型砷钒氧簇合物1-3对RhB降解效果明显,其中化合物1和2在120min的光降解时间内即可将RhB降解完全,而化合物3在210min的光降解时间内也可使RhB降解完毕,化合物4和5也对RhB表现出了较好的降解效果。经初步分析化合物1-5对RhB的降解表现出了优异的光催化性能,源于离子液体和砷钒氧簇阴离子的协同作用。上述分析充分说明化合物1-5的砷钒氧簇基离子液体具有很好的光催化应用前景。
随后采用1×10-4mol/L甲基橙溶液、1×10-4mol/L偶氮荧光桃红两种有机污染物也进行了光降解实验,研究表明化合物1-5对这两种有机污染物没有光催化作用,这说明该类离子型液体催化剂对污染物的降解具有一定的选择性。
以上所述实施例仅是为充分说明本发明而所举的较佳的实施例,其保护范围不限于此。本技术领域的技术人员在本发明基础上所作的等同替代或变换,均在本发明的保护范围之内,本发明的保护范围以权利要求书为准。
- 还没有人留言评论。精彩留言会获得点赞!