一种非共平面大位阻聚酰亚胺气体分离膜及其制备方法与流程
本发明属于气体膜分离技术领域,涉及一种用于气体分离的高性能聚酰亚胺膜、制备方法和应用,特别涉及一种非共平面大位阻聚酰亚胺气体分离膜、及其制备方法和应用。
背景技术:
膜分离技术是近几十年来发展起来的一种高效分离、提纯和净化的新型技术,和传统分离技术(如精馏、吸收、吸附、深冷分离等)相比,具有能耗低、设备简单、灵活性高、操作安全方便、环境友好等优点,其研究和应用发展非常迅速。经过近半个世纪的发展,科研人员根据分离体系和目的的不同,已研制出了多种类型的气体分离膜,己成功应用于空气分离、氢回收、天然气中酸性气体的脱除、有机蒸汽的回收等方面。目前,我国的co2分离膜和有机蒸汽膜与国外先进水平相当,但富氧和氢膜分离性能和国外尚有一定差距。
气体膜分离技术是以气体在膜两侧的分压差为传质推动力,利用不同气体分子透过膜材料渗透速率的差异来实现组分分离。其中膜材料是膜分离技术的核心,开发新型的高性能气体分离膜材料对促进膜技术的快速发展具有重要作用,是目前膜分离研究领域的重中之重。近50年来,许多新型聚合物材料如醋酸纤维素、聚砜、聚醚砜、聚酰亚胺、聚醚酰亚胺等已被应用于气体分离膜的制备。其中含氮芳杂环聚合物膜材料兼具高透气性和高选择性,尤其以聚酰亚胺的综合性能最佳,是气体分离的理想材料。
虽然已有聚酰亚胺膜具有良好的选择性,但仍存在渗透性差同时溶解性差、难以加工成膜等问题。因此,制备一种渗透性及成膜性好、制备工艺简单、易于产业化的聚酰亚胺气体分离膜是当前亟需解决的技术问题。为改善其渗透性和溶解性,可在分子水平上设计其结构单元,通过筛选单体和合成新的二酐和二胺以及聚合反应条件的控制,制备出透气性与选择性俱佳的膜材料。
技术实现要素:
为克服上述现有技术存在的问题,本发明的目的在于提供一种分离性能好和成膜性好的聚酰亚胺气体分离膜及制备方法。
本发明采用的技术方案如下:
一种非共平面大位阻聚酰亚胺气体分离膜,其非共平面聚酰亚胺的结构如下:
特别的,ar选自
以上所述非共平面大位阻聚酰亚胺气体分离膜以4,4’-二胺基-3,3’-二甲基联苯、n-溴代琥珀酰亚胺、羟基金刚烷为单体制备具有非共平面大位阻二胺单体后,再用所述二胺与芳香二酐聚合制备得到聚酰胺酸胶液,然后采用热酰亚胺化成膜原理制备得到聚酰亚胺气体分离膜。
所述聚酰亚胺气体分离膜的制备方法,具体包括如下工艺步骤:
(1)二胺单体的合成:将4,4’-二胺基-3,3’-二甲基联苯溶于氯苯,而后加入适量n-溴代琥珀酰亚胺和偶氮二异丁腈,100~150℃下反应10~20h,倒入适量甲醇中沉淀,过滤,洗涤,干燥,得到4,4’-二胺基-3,3’-二溴甲基联苯;将1-羟基金刚烷溶于适量四氢呋喃,加入适量氢化钠,反应0.2~5h后加入适量4,4’-二胺基-3,3’-二溴甲基联苯,30~100℃下反应3~12h,倒入甲醇中沉淀,过滤,洗涤,干燥,得到具有非共平面大位阻二胺单体;
(2)聚酰胺酸的合成:在氮气保护下,将步骤(1)得到的所述二胺单体在搅拌下溶解于适量极性溶剂中,溶解完毕后,在50~100℃下分3次加入适量芳香二酐,每次间隔5min,加料完毕后继续搅拌2~12h,得到聚酰胺酸胶液,得到的所述聚酰胺酸胶液用保鲜膜密封备用;
所述极性溶剂为n,n-二甲基乙酰胺、n,n-二甲基甲酰胺、n-甲基吡咯烷酮、二甲基亚砜、环丁砜的一种或几种;所述芳香二酐为均苯四甲酸酐、联苯四羧酸二酐、联苯醚四羧酸二酐、联苯酮四羧酸二酐、六氟二酐、二甲基四羧酸二酐的一种或几种;
(3)聚酰亚胺膜的制备:将步骤(2)中所述聚酰胺酸在洗净的玻璃板上刮膜,调节涂膜的厚度在50~150μm之间,放入烘箱中采用梯度升温进行热酰亚胺化反应,热酰亚胺化温度为80~220℃,时间为5~12h,自然冷却到室温进行脱膜后得到聚酰亚胺膜。
步骤(1)中所述4,4’-二胺基-3,3’-二甲基联苯、n-溴代琥珀酰亚胺、偶氮二异丁腈的摩尔比为1:(2~4):(1~2);所述1-羟基金刚烷、氢化钠、4,4’-二胺基-3,3’-二溴甲基联苯的摩尔比为1:1:(0.3~1)。
步骤(2)中所述二胺单体和芳香二酐的摩尔比为1:(0.5~2)。
本发明制得的所述非共平面大位阻聚酰亚胺膜的h2渗透系数为20~150barrer,o2渗透系数为1~20barrer,n2渗透系数为0.2~5barrer,co2渗透系数为5~60barrer。
本发明制得的所述聚酰亚胺气体分离膜的h2/n2选择性为60~150,o2/n2选择性为5~10,co2/n2选择性为10~50。
本发明所述的非共平面大位阻聚酰亚胺膜适用于气体分离,特别适用于含氢气体中分离回收氢气,含碳、含硫气体中co2、h2s的分离和回收(如天然气中co2的脱除等),空气富氧或富氮等。
本发明所述的非共平面大位阻聚酰亚胺膜材料,侧链的两个非共平面大位阻金刚烷结构,使得本发明的聚酰亚胺具有良好的溶解性和高的渗透性,成膜性优良,加工成型容易,制备工艺简单,易于产业化,便于推广应用。
附图说明
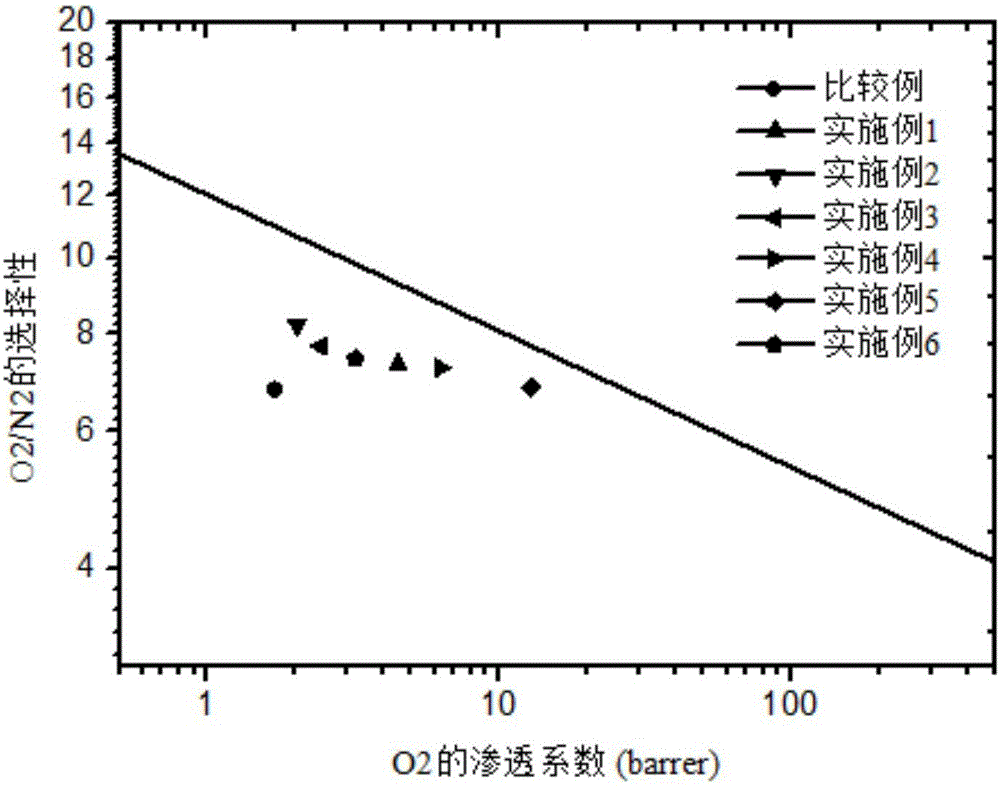
图1为o2/n2的分离上限图。
图1中直线为upperboundline(2008年),即robeson上限(2008年)。渗透系数p和选择性α是衡量气膜分离性能的两个主要参数,但分离膜的气体渗透性和分离性之间存在trade-off关系,即渗透系数的提高常会导致选择系数的降低,反之亦然,因此提高分离性能实际上是在渗透系数p和选择性α之间找到最优化的平衡点。robeson在1991年、2008年分别研究汇总了大量己发表文献中的气体分离性能数据,得到了当时某些聚合物材料能够达到的经验性分离性能上限,并将结果绘制成αij~pi的双对数坐标图,称为upperboundline,即robeson上限。
具体实施方式
下面结合附图和具体实施方式对本发明作进一步详细说明。
实施例1:
将2.123g4,4’-二胺基-3,3’-二甲基联苯溶于50ml氯苯,而后加入5.337gn-溴代琥珀酰亚胺和0.164g偶氮二异丁腈,130℃反应12h,倒入500ml甲醇中沉淀,过滤,洗涤,干燥,得到4,4’-二胺基-3,3’-二溴甲基联苯。将1.522g1-羟基金刚烷溶于50ml四氢呋喃,加入0.24g氢化钠,反应0.5h后加入1.85g4,4’-二胺基-3,3’-二溴甲基联苯,40℃反应10h,倒入甲醇中沉淀,过滤,洗涤,干燥,得到具有非共平面大位阻二胺单体。在氮气保护下,将1.709g二胺单体在搅拌下溶解于20ml极性溶剂中,溶解完毕后,在70℃下分3次加入等摩尔量的均苯四甲酸酐,每次间隔5min,加料完毕后继续搅拌3h,得到聚酰胺酸胶液,得到的所述聚酰胺酸胶液用保鲜膜密封备用。
在铺膜机底层铺上洗净的玻璃板,将模具放在玻璃板上,调节涂膜的厚度约100μm,再倒入制得的所述聚酰胺酸胶液,转动开关进行铺膜。将涂膜好的玻璃板放入烘箱中采用梯度升温进行热酰亚胺化反应,先80℃预烘2h,120℃烘1h,升温至160℃烘1h,然后升温至180℃烘1h,再升温至220℃烘1h。自然冷却到室温进行脱膜后得到聚酰亚胺膜。
该实施例制得的聚酰亚胺膜的分离性能如表1所示,其对h2的渗透系数可达55.36barrer,o2渗透系数达到4.50barrer,n2渗透系数为0.61barrer,co2渗透系数达到17.81barrer,h2/n2选择性为90.1,o2/n2选择性为7.3,co2/n2选择性为29.0。和对比例即商业化matrimid膜相比,该实施例的o2/n2和co2/n2选择性分别提高了7%、12%,同时o2、n2和co2的渗透系数分别提高了2.6、2.4、2.7倍。
实施例2:
将2.123g4,4’-二胺基-3,3’-二甲基联苯溶于50ml氯苯,而后加入5.337gn-溴代琥珀酰亚胺和0.164g偶氮二异丁腈,130℃反应12h,倒入500ml甲醇中沉淀,过滤,洗涤,干燥,得到4,4’-二胺基-3,3’-二溴甲基联苯。将1.522g1-羟基金刚烷溶于50ml四氢呋喃,加入0.24g氢化钠,反应1h后加入1.85g4,4’-二胺基-3,3’-二溴甲基联苯,50℃反应8h,倒入甲醇中沉淀,过滤,洗涤,干燥,得到具有非共平面大位阻二胺单体。在氮气保护下,将1.709g二胺单体在搅拌下溶解于20ml极性溶剂中,溶解完毕后,在80℃下分3次加入等摩尔量的联苯四羧酸二酐,每次间隔5min,加料完毕后继续搅拌5h,得到聚酰胺酸胶液,得到的所述聚酰胺酸胶液用保鲜膜密封备用。
在铺膜机底层铺上洗净的玻璃板,将模具放在玻璃板上,调节涂膜的厚度约100μm,再倒入制得的所述聚酰胺酸胶液,转动开关进行铺膜。将涂膜好的玻璃板放入烘箱中采用梯度升温进行热酰亚胺化反应,先80℃预烘2h,120℃烘1h,升温至160℃烘2h,然后升温至180℃烘0.5h,再升温至220℃烘2h。自然冷却到室温进行脱膜后得到聚酰亚胺膜。
该实施例制得的聚酰亚胺膜的分离性能如表1所示,其对h2的渗透系数可达29.70barrer,o2渗透系数达到2.03barrer,n2渗透系数为0.25barrer,co2渗透系数达到10.18barrer,h2/n2选择性为120.3,o2/n2选择性为8.2,co2/n2选择性为41.2。和对比例即商业化matrimid膜相比,该实施例的o2/n2和co2/n2选择性分别提高了21%、58%,同时o2和co2的渗透系数分别提高了1.2、1.6倍。
实施例3:
将2.123g4,4’-二胺基-3,3’-二甲基联苯溶于50ml氯苯,而后加入5.337gn-溴代琥珀酰亚胺和0.164g偶氮二异丁腈,130℃反应12h,倒入500ml甲醇中沉淀,过滤,洗涤,干燥,得到4,4’-二胺基-3,3’-二溴甲基联苯。将1.522g1-羟基金刚烷溶于50ml四氢呋喃,加入0.24g氢化钠,反应1h后加入1.85g4,4’-二胺基-3,3’-二溴甲基联苯,60℃反应6h,倒入甲醇中沉淀,过滤,洗涤,干燥,得到具有非共平面大位阻二胺单体。在氮气保护下,将1.709g二胺单体在搅拌下溶解于20ml极性溶剂中,溶解完毕后,在70℃下分3次加入等摩尔量的联苯醚四羧酸二酐,每次间隔5min,加料完毕后继续搅拌10h,得到聚酰胺酸胶液,得到的所述聚酰胺酸胶液用保鲜膜密封备用。
在铺膜机底层铺上洗净的玻璃板,将模具放在玻璃板上,调节涂膜的厚度约100μm,再倒入制得的所述聚酰胺酸胶液,转动开关进行铺膜。将涂膜好的玻璃板放入烘箱中采用梯度升温进行热酰亚胺化反应,先80℃预烘2h,120℃烘1h,升温至160℃烘1h,然后升温至180℃烘1.5h,再升温至220℃烘1.5h。自然冷却到室温进行脱膜后得到聚酰亚胺膜。
该实施例制得的聚酰亚胺膜的分离性能如表1所示,其对h2的渗透系数可达36.81barrer,o2渗透系数达到2.44barrer,n2渗透系数为0.32barrer,co2渗透系数达到11.68barrer,h2/n2选择性为116.6,o2/n2选择性为7.7,co2/n2选择性为37.0。和对比例即商业化matrimid膜相比,该实施例的o2/n2和co2/n2选择性分别提高了13%、42%,同时o2、n2和co2的渗透系数分别提高了1.4、1.3、1.8倍。
实施例4:
将2.123g4,4’-二胺基-3,3’-二甲基联苯溶于50ml氯苯,而后加入5.337gn-溴代琥珀酰亚胺和0.164g偶氮二异丁腈,130℃反应12h,倒入500ml甲醇中沉淀,过滤,洗涤,干燥,得到4,4’-二胺基-3,3’-二溴甲基联苯。将1.522g1-羟基金刚烷溶于50ml四氢呋喃,加入0.24g氢化钠,反应3h后加入1.85g4,4’-二胺基-3,3’-二溴甲基联苯,60℃反应6h,倒入甲醇中沉淀,过滤,洗涤,干燥,得到具有非共平面大位阻二胺单体。在氮气保护下,将1.709g二胺单体在搅拌下溶解于20ml极性溶剂中,溶解完毕后,在70℃下分3次加入等摩尔量的联苯酮四羧酸二酐,每次间隔5min,加料完毕后继续搅拌5h,得到聚酰胺酸胶液,得到的所述聚酰胺酸胶液用保鲜膜密封备用。
在铺膜机底层铺上洗净的玻璃板,将模具放在玻璃板上,调节涂膜的厚度约100μm,再倒入制得的所述聚酰胺酸胶液,转动开关进行铺膜。将涂膜好的玻璃板放入烘箱中采用梯度升温进行热酰亚胺化反应,先80℃预烘4h,120℃烘1h,升温至160℃烘1h,然后升温至180℃烘1h,再升温至220℃烘1h。自然冷却到室温进行脱膜后得到聚酰亚胺膜。
该实施例制得的聚酰亚胺膜的分离性能如表1所示,其对h2的渗透系数可达75.40barrer,o2渗透系数达到6.27barrer,n2渗透系数为0.87barrer,co2渗透系数达到22.83barrer,h2/n2选择性为87.0,o2/n2选择性为7.2,co2/n2选择性为26.3。和对比例即商业化matrimid膜相比,虽然该实施例的o2/n2和co2/n2选择性基本不变,但o2、n2和co2的渗透系数提高较大,分别提高了3.7、3.5、3.5倍。
实施例5:
将2.123g4,4’-二胺基-3,3’-二甲基联苯溶于50ml氯苯,而后加入5.337gn-溴代琥珀酰亚胺和0.164g偶氮二异丁腈,130℃反应12h,倒入500ml甲醇中沉淀,过滤,洗涤,干燥,得到4,4’-二胺基-3,3’-二溴甲基联苯。将1.522g1-羟基金刚烷溶于50ml四氢呋喃,加入0.24g氢化钠,反应1h后加入1.85g4,4’-二胺基-3,3’-二溴甲基联苯,70℃反应5h,倒入甲醇中沉淀,过滤,洗涤,干燥,得到具有非共平面大位阻二胺单体。在氮气保护下,将1.709g二胺单体在搅拌下溶解于20ml极性溶剂中,溶解完毕后,在70℃下分3次加入等摩尔量的六氟二酐,每次间隔5min,加料完毕后继续搅拌3h,得到聚酰胺酸胶液,得到的所述聚酰胺酸胶液用保鲜膜密封备用。
在铺膜机底层铺上洗净的玻璃板,将模具放在玻璃板上,调节涂膜的厚度约100μm,再倒入制得的所述聚酰胺酸胶液,转动开关进行铺膜。将涂膜好的玻璃板放入烘箱中采用梯度升温进行热酰亚胺化反应,先80℃预烘2h,120℃烘5h,升温至160℃烘1h,然后升温至180℃烘1h,再升温至220℃烘1h。自然冷却到室温进行脱膜后得到聚酰亚胺膜。
该实施例制得的聚酰亚胺膜的分离性能如表1所示,其对h2的渗透系数可达139.25barrer,o2渗透系数达到12.82barrer,n2渗透系数为1.87barrer,co2渗透系数达到43.98barrer,h2/n2选择性为74.3,o2/n2选择性为6.8,co2/n2选择性为23.5。和对比例即商业化matrimid膜相比,虽然该实施例的选择性有所下降,但o2、n2和co2的渗透系数有很大提高,分别提高了7.5、7.5、6.8倍。
实施例6:
将2.123g4,4’-二胺基-3,3’-二甲基联苯溶于50ml氯苯,而后加入5.337gn-溴代琥珀酰亚胺和0.164g偶氮二异丁腈,130℃反应12h,倒入500ml甲醇中沉淀,过滤,洗涤,干燥,得到4,4’-二胺基-3,3’-二溴甲基联苯。将1.522g1-羟基金刚烷溶于50ml四氢呋喃,加入0.24g氢化钠,反应1h后加入1.85g4,4’-二胺基-3,3’-二溴甲基联苯,80℃反应4h,倒入甲醇中沉淀,过滤,洗涤,干燥,得到具有非共平面大位阻二胺单体。在氮气保护下,将1.709g二胺单体在搅拌下溶解于20ml极性溶剂中,溶解完毕后,在70℃下分3次加入等摩尔量的二甲基四羧酸二酐,每次间隔5min,加料完毕后继续搅拌10h,得到聚酰胺酸胶液,得到的所述聚酰胺酸胶液用保鲜膜密封备用。
在铺膜机底层铺上洗净的玻璃板,将模具放在玻璃板上,调节涂膜的厚度约100μm,再倒入制得的所述聚酰胺酸胶液,转动开关进行铺膜。将涂膜好的玻璃板放入烘箱中采用梯度升温进行热酰亚胺化反应,先80℃预烘3h,120℃烘1h,升温至160℃烘2h,然后升温至180℃烘1h,再升温至220℃烘1h。自然冷却到室温进行脱膜后得到聚酰亚胺膜。
该实施例制得的聚酰亚胺膜的分离性能如表1所示,其对h2的渗透系数可达46.78barrer,o2渗透系数达到3.22barrer,n2渗透系数为0.43barrer,co2渗透系数达到13.77barrer,h2/n2选择性为108.0,o2/n2选择性为7.4,co2/n2选择性为31.8。和对比例即商业化matrimid膜相比,该实施例的o2/n2和co2/n2选择性分别提高了9%、22%,同时o2、n2和co2的渗透系数分别提高了1.9、1.7、2.1倍。
对比例:
选取文献(journalmembranescience,2003,225,77-90)中报导的matrimid膜做对比例,将本发明实施例1~6的非共平面大位阻聚酰亚胺膜与其比较,详细性能如表1所示。
表1本发明非共平面大位阻聚酰亚胺膜和商业化膜的分离性能比较
从表1可以看出,本发明的非共平面大位阻聚酰亚胺气体分离膜的h2渗透系数为20~150barrer,o2渗透系数为1~20barrer,n2渗透系数为0.2~5barrer,co2渗透系数为5~60barrer。分离膜的h2/n2选择性为60~150,o2/n2选择性为5~10,co2/n2选择性为10~50。
本发明的非共平面大位阻聚酰亚胺膜在保持选择性和商业化matrimid膜差不多甚至更高的情况下,其渗透系数大幅提高,o2的渗透系数增加了约1.2~7.5倍,n2的渗透系数增加了约1~7.5倍,co2的渗透系数增加了约1.6~6.8倍。
从图1实施例1~6和对比例的o2/n2分离上限图中可以看出,本发明实施例1~6的非共平面大位阻聚酰亚胺膜比matrimid膜更靠近robeson上限,分离性能具有显著性的提升。
本发明的一种非共平面大位阻聚酰亚胺气体分离膜及其制备方法不局限于上述各实施例,凡采用等同替换形成的技术方案,均落在本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种ODPA型双酚A四胺支化聚酰亚胺树脂薄膜及其制备方法与流程
- 一种BTDA型双酚A四胺支化聚酰亚胺树脂薄膜及其制备方法与流程
- 一种BPDA型双酚A四胺支化聚酰亚胺树脂薄膜及其制备方法与流程
- 含非等活性三胺单体的超支化聚酰亚胺的制备及应用的制作方法与工艺
- 二胺化合物、聚酰亚胺、光学薄膜及其制备方法与流程
- 一种聚酰亚胺导电纸的制备方法与流程
- 一种超支化的荧光脂肪族聚酰胺酰亚胺及其制备方法与用途与流程
- 一种含卟啉结构的超支化聚酰亚胺的四胺单体及其聚合物与制备方法和应用与流程
- 聚(酰胺酸)合成及至高分子量聚酰亚胺的转化的制造方法与工艺
- 聚酰亚胺及/或聚酰胺酰亚胺多孔质体以及其制造方法、进行分离及/或吸附的方法、分离材料、吸附材料、过滤介质、层叠体、以及过滤装置与流程