一种聚芳醚类共价有机骨架材料及其制备方法与流程
本发明属于共价有机骨架材料技术领域,具体涉及一种高化学稳定性的聚芳醚类共价有机骨架材料及其制备方法。
背景技术
共价有机骨架材料(covalentorganicframeworks)是一种由可逆共价键连接形成的有机结晶多孔聚合物。共价有机骨架材料具有孔道规则、密度低、比表面积高、稳定性较好等特点,在气体存储与分离、催化、光电等方面具有广泛的潜在应用。
目前共价有机骨架材料的种类还非常有限(主要包括硼酸类、席夫碱类、聚酰亚胺类等),而且大多数共价有机骨架材料的化学稳定性较差,尤其是在酸碱体系中的稳定性较差,严重的限制了共价有机骨架材料的进一步发展和应用。虽然最近报道了一些相对稳定的共价有机骨架材料,但这些材料通常是通过氢键、疏水相互作用或互变异构等方式来提高化学稳定性,但是结构中仍然存在弱键,致使其化学稳定性依然受到限制。
技术实现要素:
为了克服现有材料中存在的上述不足,本发明利用芳香亲核取代反应,合成了一系列基于聚芳醚结构的共价有机骨架材料,并通过多种手段进行表征。这种材料具有极高的化学稳定性,可以在沸水、强酸、强碱、强氧化剂、强还原剂和大多数有机溶剂中保持其结构。其稳定性超越了已知的大多数结晶多孔材料,包括分子筛、金属有机骨架材料及其它共价有机骨架材料。这种材料具有优越的二氧化碳吸附与分离性能。同时该材料可以进行多种后修饰,可以在保持其骨架和孔道结构的前提下获得多种官能团修饰的结构。
本发明通过如下技术方案实现:
一种聚芳醚类共价有机骨架材料,该材料的结构式如下:
其中,r为氰基、硝基、羧基、酯基、醛基或磺基;
或
本发明的另一目的在于提供一种聚芳醚类共价有机骨架材料的制备方法,具体步骤如下:
步骤(1):取1~100当量无水碳酸钾加入反应管中,再取a类和b类单体按照摩尔比为1:2~2:1混合均匀,加入反应管中;所述a类反应单体为多酚单体,所述b类反应单体为含氟芳香单体或含氯芳香单体;
步骤(2):然后将1-甲基吡咯烷酮和均三甲苯按体积比1:4~4:1混合加入反应管中;液氮冷冻-抽真空-解冻,循环三次,抽真空后熔封反应管,120℃~160℃反应3~7天;
步骤(3):反应结束后分别用n,n-二甲基甲酰胺、水、四氢呋喃依次洗涤,用丙酮溶剂交换,抽滤,然后真空干燥得到聚芳醚类共价有机骨架材料。
进一步地,所述的a类反应单体为2,3,6,7,10,11-六羟基三亚苯或1,2,4,5-四羟基苯。
进一步地,所述的b类反应单体为四氟对苯二乙腈,2,3,5,6-四氟吡啶-4-腈,2,3,6,7-四氟蒽醌,2,3,8,9,14,15-六氟-5,6,11,12,17,18-六氮杂三萘或2,3,8,9,14,15-六氯-5,6,11,12,17,18-六氮杂三萘。
与现有技术相比,本发明的优点如下:
1、产物具有极高的化学稳定性,可以在沸水、强酸、强碱、强氧化剂、强还原剂和大多数有机溶剂中保持其结构;
2、产物具有高的结晶度和比表面积;
3、产物具有高的二氧化碳吸附及分离性能;
4、产物可以在多种条件下进行后修饰,在保持骨架和孔道结构的情况下得到不同官能团修饰的孔道和表面;
附图说明
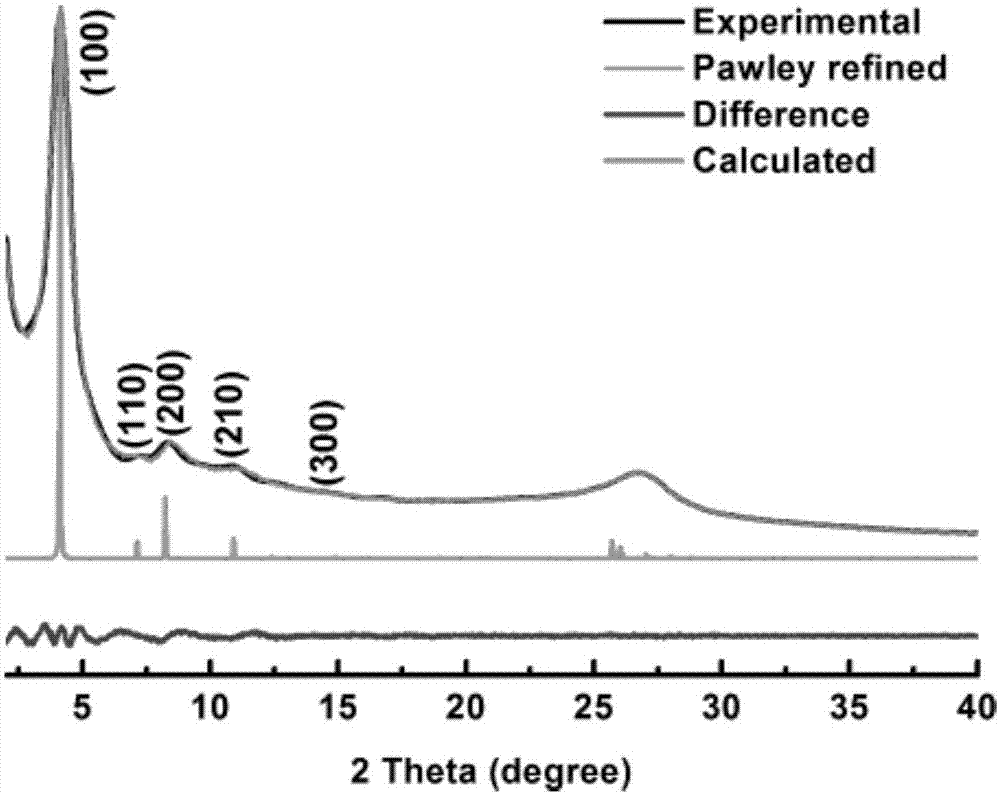
图1为本发明实施例1制备的共价有机骨架材料的x射线衍射图;
图2为本发明实施例1制备的共价有机骨架材料的氮气吸附等温线图;
图3为本发明实施例1制备的共价有机骨架材料的扫描电子显微镜图;
图4为本发明实施例1制备的共价有机骨架材料的红外图;
图5为本发明实施例1制备的共价有机骨架材料在氮气气氛下的热重分析谱图;
图6为本发明实施例1制备的共价有机骨架材料的固体核磁图;
图7为本发明实施例2制备的共价有机骨架材料的x射线衍射图;
图8为本发明实施例2制备的共价有机骨架材料的氮气吸附等温线图;
图9为本发明实施例2制备的共价有机骨架材料的扫描电子显微镜图;
图10为本发明实施例2制备的共价有机骨架材料的红外图;
图11为本发明实施例2制备的共价有机骨架材料在氮气气氛下的热重分析谱图;
图12为本发明实施例2制备的共价有机骨架材料的固体核磁图;
图13为本发明实施例1制备的共价有机骨架材料在不同有机溶剂处理一周后的x射线衍射图;
图14为本发明实施例1制备的共价有机骨架材料在不同溶液体系处理一周后的x射线衍射图;
图15为本发明实施例1制备的共价有机骨架材料在不同溶液体系处理一周后的红外图;
图16为本发明实施例1制备的共价有机骨架材料在273k的二氧化碳、甲烷和氮气吸附图;
图17为本发明实施例1制备的共价有机骨架材料在298k的二氧化碳、甲烷和氮气吸附图;
图18为本发明实施例5、实施例6制备的共价有机骨架材料的x射线衍射图;
图19为本发明实施例5制备的共价有机骨架材料的氮气吸附等温线图;
图20为本发明实施例5制备的共价有机骨架材料的扫描电子显微镜图;
图21为本发明实施例5、实施例6制备的共价有机骨架材料的红外图;
图22为本发明实施例5制备的共价有机骨架材料的固体核磁图;
图23为本发明实施例6制备的共价有机骨架材料的氮气吸附等温线图;
图24为本发明实施例6制备的共价有机骨架材料的扫描电子显微镜图;
图25为本发明实施例6制备的共价有机骨架材料的固体核磁图。
具体实施方式
下面结合附图对本发明做进一步地说明。
下述实施例中所述实验方法,如无特殊说明,均为常规方法;所述试剂和材料,如无特殊说明,均可从商业途径获得。
实施例1共价有机骨架材料juc-505的制备与表征
称取无水碳酸钾138.2mg于反应管中,称取2,3,6,7,10,11-六羟基三亚苯32.4mg和四氟对苯二乙腈30.0mg,研磨均匀,将混合物加入反应管中,加入均三甲苯0.3ml与1-甲基吡咯烷酮0.6ml,液氮冷冻-抽真空-融化,循环三次,抽真空后熔封反应管,120℃反应3天。反应结束后依次用n,n-二甲基甲酰胺,水和四氢呋喃充分浸泡洗涤,用丙酮溶剂交换,抽滤,然后真空干燥得到褐色粉末,结构式如下:
其产物xrd及按aa堆积模拟粉末xrd衍射图谱如图1,可见模拟谱图和产物谱图是吻合的。
其产物在77k的氮气吸附等温线如图2,可见其呈现典型的ⅰ型吸附等温线,说明产物为微孔结构。其孔隙率为0.51cm3g-1,bet比表面积为1584m2g-1,根据非定域密度泛函理论计算得到其孔径为
其产物扫描电镜照片如图3,可见所合成产物为球状晶体的聚集体。
其产物的红外图谱如图4,从图可见产物在1269cm-1和1021cm-1附近有较强吸收,分别对应于c-o-c的不对称和对称振动,说明单体发生了缩合反应;产物在2239cm-1有较强吸收,对应于c≡n的振动,说明共价有机骨架材料中仍然有大量氰基。
其产物在氮气气氛的热重图谱如图5,气体流速30ml/min,升温速率10℃/min,其在500℃左右有一个明显的失重峰,说明骨架在氮气气氛下可以稳定到500℃。
其产物的固体核磁图谱如图6。从图可见所得产物的化学位移与结构有较好的对应关系。
实施例2共价有机骨架材料juc-506的制备与表征
称取无水碳酸钾138.2mg于反应管中,称取2,3,6,7,10,11-六羟基三亚苯32.4mg和,3,6,7-四氟蒽醌42.0mg,研磨均匀,将混合物加入反应管中,加入均三甲苯0.3ml与1-甲基吡咯烷酮0.6ml,液氮冷冻-抽真空-融化,循环三次,抽真空后熔封反应管,160℃反应3天。反应结束后用n,n-二甲基甲酰胺,水和四氢呋喃洗涤,用丙酮溶剂交换,抽滤,然后真空干燥得到褐色粉末。
其产物结构式如下:
其产物xrd及按aa堆积模拟粉末xrd衍射图谱如图7,可见模拟谱图和产物谱图是吻合的。
其产物在77k的氮气吸附等温线如图8,可见其呈现典型的ⅳ型吸附等温线,说明产物为介孔结构。其孔隙率为0.71cm3g-1,bet比表面积为1655m2g-1,根据非定域密度泛函理论计算得到其孔径为
其产物扫描电镜照片如图9,可见所合成产物为球状晶体的聚集体。
其产物的红外图谱如图10,从图可见产物在1269cm-1和1021cm-1附近有较强吸收,分别对应于c-o-c的不对称和对称振动,说明单体发生了缩合反应;产物在1676cm-1有较强吸收,对应于c=o的振动,说明共价有机骨架材料中仍然有大量羰基。
其产物在氮气气氛的热重图谱如图11,气体流速30ml/min,升温速率10℃/min,其在500℃左右有一个明显的失重峰,说明骨架在氮气气氛下可以稳定到500℃。
其产物的固体核磁图谱如图12。从图可见所得产物的化学位移与结构有较好的对应关系。
实施例3juc-505的化学稳定性测试
称取实施例1中得到褐色粉末10mg,分别加入20mln,n-二甲基甲酰胺,二甲基亚砜,四氢呋喃,正己烷,甲醇,沸水(120℃),浓盐酸(12m),浓硫酸(18m),氢氟酸(40%),饱和氢氧化钠(14m),甲醇钠(5m甲醇溶液),铬酸洗液(0.5g重铬酸钾溶于2ml水和18ml浓硫酸)和氢化铝锂(2.4m四氢呋喃溶液),浸泡一周。样品用8m盐酸(对于氢化铝锂浸泡的样品),水(对于水溶液处理的样品),甲醇(对于甲醇钠浸泡的样品)和四氢呋喃洗涤,随后用丙酮进行溶剂交换,85℃干燥12小时后进行粉末x射线衍射测试和红外测试。
其xrd谱图如图13,14,从图可见样品在所有体系下都保持了其晶体结构。
其红外谱图如图15,从图可见样品在所有体系下都保持了其醚键结构。
实施例4juc-505的二氧化碳吸附与分离性能测试
称取实施例1中得到褐色粉末50mg,在273k和298k测试其二氧化碳、甲烷和氮气的气体吸附,结果如图16,17。其在273k时二氧化碳吸附量231.3mgg-1,甲烷吸附量19.2mgg-1,氮气吸附量10.4mgg-1,根据亨利区斜率计算得到的二氧化碳/甲烷和二氧化碳/氮气理想吸附选择性分别为48.85和114.23;其在298k时二氧化碳吸附量189.0mgg-1,甲烷吸附量15.2mgg-1,氮气吸附量8.8mgg-1,根据亨利区斜率计算得到的二氧化碳/甲烷和二氧化碳/氮气理想吸附选择性分别为33.79和65.59。
实施例5juc-505的羧基后修饰制备juc-505-cooh
称取实施例1中得到褐色粉末200mg于带回流冷凝管的反应瓶中,加入20%氢氧化钠溶液(乙醇:水=1:1)50ml,120℃回流3天。反应体系冷却至室温,抽滤。固体用去离子水和1m盐酸回流2小时,冷却至室温后抽滤,水洗至中性后用四氢呋喃洗。用四氢呋喃索氏提取一天后用无水丙酮交换溶剂。80℃真空干燥得到褐色粉末。
其产物xrd及juc-505的xrd衍射图谱如图18,可见其在修饰前后骨架保持稳定。
其产物在77k的氮气吸附等温线如图19,可见其呈现典型的ⅰ型吸附等温线,说明产物为微孔结构。其bet比表面积为1392m2g-1,说明在修饰后仍然保持其孔道结构。
其产物扫描电镜照片如图20,可见在修饰后其形貌没有明显变化。
其产物的红外图谱如图21,从图可见产物c-o-c的振动没有明显变化,说明在修饰后骨架保持不变;产物在1716cm-1有较强吸收,对应于羧基的振动,同时2239cm-1的氰基振动彻底消失,说明在修饰后氰基转化为羧基。
其产物的固体核磁图谱如图22。从图可见所得产物的化学位移与结构有较好的对应关系,163ppm处的峰对应于羧基碳的化学位移。
实施例6juc-505的氨基后修饰制备juc-505-nh2
称取实施例1中得到褐色粉末200mg于带回流冷凝管的反应瓶中,加入2.4m氢化铝锂-四氢呋喃溶液30ml,90℃回流3天。反应体系冷却至室温,淬灭后加入8m盐酸10ml,继续搅拌1小时。抽滤,水洗。固体加10%氢氧化钠20ml除去过量的酸,用水洗至中性后用四氢呋喃洗。用四氢呋喃索氏提取一天后用无水丙酮交换溶剂。80℃真空干燥得到褐色粉末。
其产物xrd及juc-505的xrd衍射图谱如图18,可见其在修饰前后骨架保持稳定。
其产物在77k的氮气吸附等温线如图23,可见其呈现典型的ⅰ型吸附等温线,说明产物为微孔结构。其bet比表面积为1244m2g-1,说明在修饰后仍然保持其孔道结构。
其产物扫描电镜照片如图24,可见在修饰后其形貌没有明显变化。
其产物的红外图谱如图21,从图可见产物c-o-c的振动没有明显变化,说明在修饰后骨架保持不变;产物在3293cm-1有较强吸收,对应于氨基的振动,同时2239cm-1的氰基振动彻底消失,说明在修饰后氰基转化为氨基。
其产物的固体核磁图谱如图25。从图可见所得产物的化学位移与结构有较好的对应关系,25ppm处的峰对应于氨甲基碳的化学位移。
以上所述仅为本发明的优选实施例,并不能作为限制本发明的依据,对于本领域的技术研究人员来说,可以根据本发明的实施例对技术方案进行修改、等同替换、改进等,而所有这些变动都应属于本发明权利要求的保护范围之内。
综上,本发明利用酚和芳香氟化物的亲核取代反应,在溶剂热条件下合成了包括一系列聚芳醚类新型共价有机骨架材料,这种材料具有超高的化学稳定性,在沸水、强酸、强碱、强氧化剂、强还原剂和大多数有机溶剂中均能保持其结构。其稳定性超过了大多数已知的结晶多孔材料,包括分子筛,金属有机骨架材料和其他共价有机骨架材料。这种材料具有优越的二氧化碳吸附与分离性能。同时该材料可以进行多种后修饰,可以在保持其骨架和孔道结构的前提下获得多种官能团修饰的结构。
- 还没有人留言评论。精彩留言会获得点赞!