一种快速检测百草枯和/或敌草快的方法及试剂盒与流程
本发明属于分析化学领域,涉及一种快速检测百草枯和/或敌草快的方法,特别是一种不同基质生物样品中百草枯和/或敌草快含量的表面增强拉曼光谱分析方法。
背景技术:
百草枯(Paraquat,PQ),又称对草快、克芜踪(Gramoxone),化学名为1,1-二甲基-4,4’-联吡啶阳离子盐,是一种使用广泛的季铵盐类除草剂,对人毒性极大。自1962年首次使用以来,百草枯自杀、投毒事件屡屡发生,因无特效解毒剂,临床救治难度很大,口服中毒者死亡率在90%以上。百草枯吸收后经血液循环分布于血液、全身器官,不经代谢多以原型从肾脏排出,故血浆和尿液常作为检测中毒患者体内百草枯的常用检材。
目前,针对百草枯中毒血、尿样本的检测方法主要有气相色谱法、高效液相色谱法、质谱联用法、分光光度法、酶联免疫法等,大部分方法前处理过程复杂或分析时间较长。
表面增强拉曼光谱(Surface-Enhanced Raman Spectroscopy,简称SERS)技术是一种指纹光谱技术,具有高灵敏度、高分辨率、水干扰小、稳定性好等优点,适合痕量分析。目前,虽已有用SERS方法检测水体、食品中百草枯残留,但均不适用于生物样品基质。
因而,需要建立快速、灵敏、准确地检测百草枯中毒患者生物样品,这对患者的及时救治至关重要。
技术实现要素:
本发明的目的是克服现有技术的缺陷和不足,提供一种快速检测 百草枯和/或敌草快的方法。发明人令人惊讶地发现,I-离子、S2-离子或F-离子与金纳米粒子和百草枯或敌草快之间均具有很好作用力,I-离子、S2-离子或F-离子可将被分析物百草枯或敌草快吸附于金纳米粒子的表面,从而大幅增强其拉曼峰强度和特征性。I-好比一座“桥梁”架于百草枯或敌草快和Au核之间,不仅能够大幅增强其拉曼峰强度和特征性,还能够清除金纳米粒子表面吸附的蛋白等基质,从而有效抑制蛋白等基质对百草枯和/或敌草快检测的干扰。
基于上述发现,本发明涉及以下几个方面。
本发明的第一方面涉及一种试剂盒,其中包含:金纳米粒子和能够提供I-离子、S2-离子或F-离子的物质。其中金纳米粒子和I-离子、S2-离子或F-离子的摩尔比为1:4×103~8×105,优选为1:2×104~6×105,进一步优选1:4×104~4.8×105,更进一步优选1:8×104~4.8×105,再进一步优选1:2×105~4.8×105,特别优选1:4×105。
本发明的第二方面涉及本发明所述的试剂盒用于检测样品中百草枯和/或敌草快的用途。其中金纳米粒子和I-离子、S2-离子或F-离子的摩尔比为1:4×103~8×105,优选为1:2×104~6×105,进一步优选1:4×104~4.8×105,更进一步优选1:8×104~4.8×105,再进一步优选1:2×105~4.8×105,特别优选1:4×105。
本发明的第三方面涉及一种检测样品中百草枯和/或敌草快的方法,该方法使用本发明所述的试剂盒对样品进行处理,采用表面增强拉曼光谱分析法进行检测。
本发明的第四方面涉及金纳米粒子和能够提供I-离子、S2-离子或F-离子的物质在制备用于检测样品中百草枯和/或敌草快的试剂盒中的用途。其中金纳米粒子和I-离子、S2-离子或F-离子的摩尔比为1:4×103~8×105,优选为1:2×104~6×105,进一步优选1:4×104~4.8×105,更进一步优选1:8×104~4.8×105,再进一步优选1:2×105~4.8×105,特别优选1:4×105。
本发明的第五方面涉及金纳米粒子和能够提供I-离子、S2-离子或F-离子的物质联合用于检测样品中百草枯和/或敌草快的用途。其中金 纳米粒子和I-离子、S2-离子或F-离子的摩尔比为1:4×103~8×105,优选为1:2×104~6×105,进一步优选1:4×104~4.8×105,更进一步优选1:8×104~4.8×105,再进一步优选1:2×105~4.8×105,特别优选1:4×105。
本发明第六方面涉及一种检测样品中百草枯和/或敌草快的方法,该方法包括将样品(所述的样品为未经预处理的待测样品或经过预处理的待测样品)与金纳米粒子和能够提供I-离子、S2-离子或F-离子的物质进行混合的步骤,采用表面增强拉曼光谱分析法进行检测。金纳米粒子和I-离子、S2-离子或F-离子的摩尔比为1:4×103~8×105,优选为1:2×104~6×105,进一步优选1:4×104~4.8×105,更进一步优选1:8×104~4.8×105,再进一步优选1:2×105~4.8×105,特别优选1:4×105。
发明详述
本发明中,所述的能够提供I-离子、S2-离子或F-离子的物质优选为包含I-离子、S2-离子或F-离子的盐,进一步优选为包含I-离子、S2-离子或F-离子的无机盐,特别优选为含I-离子的无机盐,例如KI、NaI、MgI2、CuI2或FeI2等。
在一个优选的实施方案中,本发明所述的试剂盒中,所述的能够提供I-离子、S2-离子或F-离子的盐可以以溶液形式存在,也可以以固体形式存在。当以固体形式存在时,使用时可将其直接加入到液体待测样品中,也可以用水溶解后再使用。
在另一个优选的实施方案中,本发明所述的试剂盒中,所述的能够提供I-离子、S2-离子或F-离子的物质也可以是能够制备I-离子、S2-离子或F-离子的化学原料,使用时,先将原料混合后反应制备得到含有I-离子、S2-离子或F-离子的物质。
本发明中,所述的金纳米粒子为金的微小颗粒,其直径在1~100nm之间,优选为约30~70nm,进一步优选为约40~60nm,更优选为50~55nm。所述的金纳米粒子可以为裸金纳米粒子,也可以为核壳型金纳米粒子,优选为有孔核壳型金纳米粒子,例如在裸金纳米粒子表面包裹SiO2壳层形成的核壳型金纳米粒子,可参照公开J.F.Li,Y. F.Huang,Y.Ding,Z.L.Yang,S.B.Li,X.S.Zhou,F.R.Fan,W.Zhang,Z.Y.Zhou,D.Y.Wu,B.Ren,Z.L.Wang,Z.Q.Tian,Nature,2010,464,392.的方法制备),优选为核壳型金纳米粒子。所述的裸金纳米粒子可以通过本领域常用的制备方法制备得到,例如,由氯金酸通过还原法制备各种不同粒径的纳米金,包括但不限于以下还原法:PVP保护还原法(兰新哲,金志浩,PVP保护还原法制备金纳米粒子,稀有金属材料与工程,2003,32(1):50-53)、磷钼酸光催化还原剂法(王振平,吴莹,牛彩红等,磷钼酸作为光催化还原剂制备金纳米粒子,光谱实验室,2007,第3期,334-337页)、柠檬酸三钠还原法(G.Frens,Nature Phys.Sci.1973,241,20.)、紫外光引发还原法(阮伟东,吕志成,纪楠等,紫外光引发还原制备金溶胶及其SERS活性的研究,光散射学报,2005,17(14):329-331)、溶胶凝胶-模板法(邵桂妮,张兴堂,刘冰等,溶胶凝胶-模板法制备一维纳米金材料,现代化工,2006,26(1):44-46)等。优选采用柠檬酸三钠还原法制备。
在一个优选的实施方案中,本发明所述的试剂盒中,所述的金纳米粒子可以以原液或浓缩后再进行检测。
本发明所述的试剂盒中,还可以包含一个或多个微量滴定板(例如96孔板)、样品预处理所需物品和/或使用说明书。所述的样品预处理所需物品包括但不限于选自以下物质中的一种或多种:固相萃取柱、分析纯甲醇、甲醇水溶液(优选的浓度为40~60%(v/v),例如50%(v/v))、三氟乙酸水溶液(优选的浓度为1~3%(v/v),例如2%(v/v))。所述的固相萃取柱中的填料优选为柱填料为弱阳离子类型,进一步优选为Cleanert PWCX型固相萃取柱。
本发明所述的试剂盒,其中所述的金纳米粒子与能够提供I-离子、S2-离子或F-离子的物质被合适地包装,可分别包装在小瓶、小袋和/或任何适用于检测方法的容器中。如果有,所述的样品预处理所需物品也分别包装在小瓶、小袋和/或任何适用于检测方法的容器中。
本发明所述的试剂盒可以用于定量或定性检测样品中的百草枯和 /或敌草快,检测时优选采用表面增强拉曼光谱分析法进行检测。
本发明中,金纳米粒子和I-离子、S2-离子或F-离子的摩尔比为1:4×103~8×105,优选为1:2×104~6×105,进一步优选1:4×104~4.8×105,更进一步优选1:8×104~4.8×105,再进一步优选1:2×105~4.8×105,特别优选1:4×105。
本发明中,所述的样品为水体、土壤、蔬菜、生肉(例如猪肉、牛肉、羊肉、马肉、驴肉等)或哺乳动物的生物样品(例如全血、血浆、体液(例如灌胃液)、尿、组织(例如肺组织)等样品)。所述的哺乳动物包括但不限于人、大鼠、小鼠、猫、狗、猪、牛、羊、马、驴、兔等等。
在一个优选的实施方案中,本发明第三方面或第六方面所述的检测样品中百草枯和/或敌草快的方法,包括以下步骤:
a)取适量样品(所述的样品为未经预处理的待测样品或经过预处理的待测样品),加入金纳米粒子,再加入适量能够提供I-离子、S2-离子或F-离子的物质可以交换,混匀,得到混合溶液,
优选地,所述的混合溶液中:金纳米粒子的浓度为0.25nmol/L,
优选地,所述的混合溶液中:I-离子、S2-离子或F-离子的浓度为1×103~2×105nmol/L,优选5×103~1.5×105nmol/L,进一步优选1×104~1.2×105nmol/L,更进一步优选2×104~1.2×105nmol/L,再进一步优选5×104~1.2×105nmol/L,特别优选1×105nmol/L。
b)取适量混合溶液置于微量滴定板中,进行拉曼光谱测试,得到百草枯和/或敌草快的拉曼谱图;
c)根据样品的特征拉曼峰强度判定百草枯的有无及其浓度。
该方法可以采用内标法或外标法进行定量。在一个优选的技术方案中,该方法采用外标法进行定量。绘制工作曲线的步骤如下:配制不同浓度的百草枯和/或敌草快标准溶液,按上述方法,测得不同浓度百草枯和/或敌草快标准溶液的拉曼信号强度,根据信号强度和浓度绘制工作曲线。
百草枯的特征拉曼峰为839cm-1、1188cm-1、1293cm-1和1643 cm-1。其中839cm-1和1643cm-1归属为C-N伸缩振动和C=N伸缩振,1188cm-1和1293cm-1归属为苯环骨架振动和联苯类CC桥键伸缩振动。本发明中采用1643cm-1作为百草枯定量或定性的特征拉曼峰,采用1570cm-1作为敌草快定量或定性的特征拉曼峰。因此,在定性或定量时,百草枯的特征拉曼峰位于1643cm-1处,敌草快的特征拉曼峰位于1570cm-1处。
当使用本发明所述的检测方法对样品中百草枯和/或敌草快进行检测时,根据样品的不同,决定是否需要进行预处理,以及采用那种方法进行预处理。
当样品为土壤、蔬菜或生肉时,用水或其他合适的溶剂提取过滤后,将滤液经过固相萃取柱(优选为柱填料为弱阳离子类型固相萃取柱,进一步优选为Cleanert PWCX型固相萃取柱)进行纯化,纯化过程为:先用甲醇和水活化固相萃取柱(甲醇和水的用量分别优选为0.5~2ml,例如1ml),上样,再用甲醇水溶液(甲醇水溶液的浓度优选为为40~60%,例如50%,用量优选为1~3ml,例如2ml)淋洗去除杂质,最后用三氟乙酸水溶液(三氟乙酸水溶液优选的浓度为1~3%,例如2%,用量优选为1~3ml,例如2ml)洗脱,得到洗脱液,即为待测样品。
当样品为水体样品、血浆或体液(例如灌胃液)样品时,无需进行预处理。
当样品为全血样品时,将全血样品离心后,弃去下层,取上层血浆层作为待测样品。
当样品为组织样品(例如肺组织)或尿液样品时,将样品经过固相萃取柱(优选为柱填料为弱阳离子类型固相萃取柱,进一步优选为Cleanert PWCX型固相萃取柱)进行纯化,纯化过程为:先用甲醇和水活化固相萃取柱(甲醇和水的用量分别优选为0.5~2ml,例如1ml),上样,再用甲醇水溶液淋洗去除杂质(甲醇水溶液的浓度优选为为40~60%(v/v),例如50%(v/v),用量优选为1~3ml,例如2ml),最后用洗脱(三氟乙酸水溶液优选的浓度为1~3%(v/v),例如2%(v/v),用 量优选为1~3ml,例如2ml),得到洗脱液,即为待测样品。
本发明的检测样品中百草枯和/或敌草快的方法的检测流程图如图1所示。
本发明所述的检测样品中百草枯和/或敌草快的方法,优选采用便携式拉曼光谱仪进行检测,激光功率为30-300mW,优选激光功率为150mW,检测方式优选为湿法。
本发明所述的检测可以用于诊断目的,也可以用于非诊断目的。用于非诊断目的时,可根据检测结果判断水体、土壤、蔬菜、生肉或其他生物组织中的百草枯和/或敌草快的污染情况,也可以判断生物体内是否含有百草枯和/或敌草快的残留。用于诊断目的时,可以满足临床百草枯和/或敌草快中毒的快速诊断。
本发明的有益技术效果
本发明利用以核/壳型纳米金纳米粒子为基底,I-离子、S2-离子或F-离子,特别是I-离子与金纳米粒子和百草枯和/或敌草快之间的特异性作用力,有效抑制生物样品中复杂基质的干扰,增强拉曼检测热点,大幅增强百草枯的特征峰强度,进而建立了一种样品中百草枯和/或敌草快含量的检测方法。加入I-离子后,一方面I-离子与金粒子和百草枯(或敌草快)之间均具有很好作用力,将被分析物百草枯(或敌草快)吸附于金纳米粒子表面,从而大幅增强其拉曼峰强度和特征性,I-离子好比一座“桥梁”架于百草枯(或敌草快)和金纳米粒子核之间;另一方面I-离子可清除金纳米粒子核表面吸附的蛋白等基质,从而有效抑制其对百草枯(或敌草快)检测的干扰。该方法能够得到清晰分明的百草枯(或敌草快)特征拉曼谱图。
该方法采用表面增强拉曼光谱分析方法,能够快速检测样品中百草枯和/或敌草快含量,适合多种样品中的百草枯和/或敌草快的检测,特别适用于哺乳动物全血、血浆、体液、尿样、肺组织样品等不同基质中的百草枯和/或敌草快含量测定,具有灵敏度高、操作简便和检测速度快等优点,并且在1小时内,该方法稳定性良好,可满足临床百草 枯和/或敌草快中毒的快速诊断。
该方法对百草枯的检测与常规液质联用(LC-MS)的检测灵敏度相当,且样品制备简单,对于血液样品其检测分析可在5min内完成;尿液和组织样品可在15min内完成。该方法对于血浆样品中的百草枯含量的检测范围为1-40μg/L,线性相关系数R2=0.983,检出限为1μg/L;对于尿液样品中的百草枯的检测范围为5-100μg/L,线性相关系数R2=0.992,检出限为4μg/L。
附图说明
图1百草枯中毒生物样品SERS检测流程图;
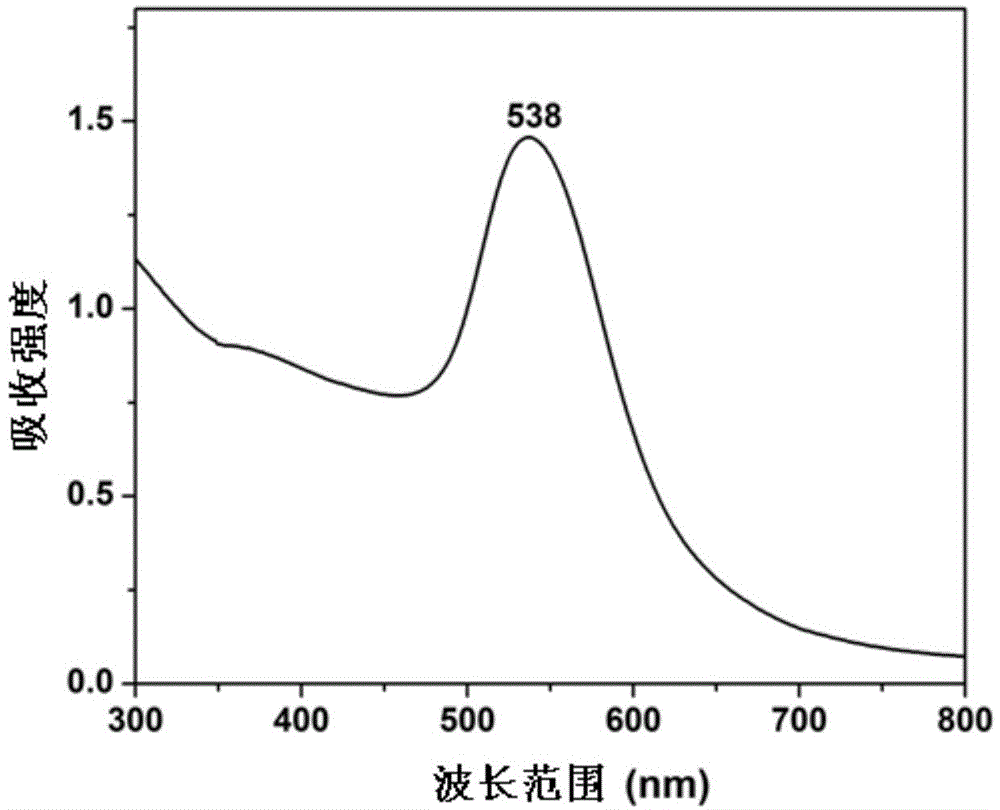
图2-A为核/壳型纳米粒子的紫外(UV-Vis)表征图;
图2-B为核/壳型纳米粒子透射电镜(TEM)表征图;
图3-A为不同的阴离子对于50μg/L百草枯样品1643cm-1处特征峰的信号影响;
图3-B为不同浓度KI(0.001、0.005、0.01、0.02、0.05、0.1、0.12、0.15、0.2mol/L)对50μg/L血浆百草枯在1643cm-1处拉曼信号强度的影响;
图4为50μg/L百草枯血浆样品不加与加入0.1mol/L KI的SERS谱图,其中a为不加KI时,b为加入KI时;
图5-A为不同浓度的百草枯血浆样品的SERS谱图;
图5-B为百草枯血浆样品在1643cm-1处的峰强-浓度工作曲线;
图6为百草枯尿样经固相萃取后,取萃取液不加与加入0.1mol/L KI的SERS谱图,其中a为不加KI时,b为加入KI时;
图7-A为不同浓度百草枯尿液样品固相萃取的SERS谱图;
图7-B为百草枯尿液样品1643cm-1处峰强-浓度工作曲线;
图8-A为血浆样品中加入百草枯、4,4'-联吡啶、敌草快不同情况得到的SERS谱图,其中:a为血浆中加入4,4'-联吡啶+百草枯+敌草快的样品加入KI时,b为血浆中加入敌草快+4,4'-联吡啶加入KI时;c为血浆中加入百草枯+4,4'-联吡啶加入KI时;d为血浆中加入4,4'- 联吡啶+百草枯+敌草快不加入KI时;
图8-B为血浆中加入敌草快+百草枯得到的SERS谱图,其中:a为加入KI时,b为不加KI时;
图9为生理盐水50μg/L百草枯中毒样品的SERS谱图;
图10为32μg/L百草枯全血样品的SERS谱图;
图11为百草枯中毒人员肺样品固相萃取液的SERS谱图;
图12为50μg/L百草枯血浆样品SERS信号随时间的变化图。
具体实施方式
下面将结合实施例对本发明的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限定本发明的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。
实施例1金纳米粒子的制备
a)裸金纳米粒子的制备
采用柠檬酸钠还原四氯金酸法制备粒径约为50nm的金纳米粒子。
准确称取100mg三水合四氯金酸于100ml容量瓶定容得1mg/mL母液,取10ml母液稀释至100ml,倒入250ml三颈瓶中油浴,在快速搅拌下加热至沸,待冷凝回流后迅速加入0.85ml 0.1%柠檬酸三钠水溶液,继续加热搅拌40min,制备完成后储存于4℃冰箱,得到裸金纳米粒子,浓度约为0.05nmol/L。制备得到的金纳米离子经过紫外光谱和透射电镜,观察粒径大小和及其均匀性,粒径约为50nm(参考文献:G.Frens,Nature Phys.Sci.1973,241,20.)
b)核壳型金纳米粒子的制备
向步骤a)中制备好的直径为50nm金纳米粒子(30ml)中加入0.4ml浓度为1mmol/L的氨丙基三甲氧基硅烷(APTMS)溶液,并搅拌15min,再加入3.2ml浓度为0.54wt%的硅酸钠溶液并搅拌3 min,使金纳米粒子表面包覆生成SiO2壳层。
当硅酸钠溶液的pH值小于10.2时,生成无孔且厚度大于2nm的SiO2壳层;当硅酸钠溶液的pH值大于11时,生成有孔且很薄的SiO2壳层。(参考文献:J.F.Li,Y.F.Huang,Y.Ding,Z.L.Yang,S.B.Li,X.S.Zhou,F.R.Fan,W.Zhang,Z.Y.Zhou,D.Y.Wu,B.Ren,Z.L.Wang,Z.Q.Tian,Nature,2010,464,392.)
制备得到的有孔核壳型纳米粒子浓度约为0.05nmol/L,经过紫外可见吸收光谱和透射电镜,观察粒径大小和及其均匀性。其紫外可见吸收光谱(UV-Vis)如图2-A所示,由Cary 300型紫外可见分光光度计(美国Varian公司)获得。从图2-A可以看出,金纳米粒子的吸收波长为530-533nm左右,直径为55nm。其纳米粒子形貌底形貌如图2-B所示,通过Tecnai-F30型高分辨率透射电镜(HR-TEM,美国FEI公司)表征,可以观察到裸金的外层包裹一层SiO2壳。
下面实施例中用到的金纳米粒子即为实施例1制备的有孔核壳型金纳米粒子。下列每个实施例中用到的离心浓缩后的有孔核壳型金纳米粒子通过下列方法获得:取实施例1制备的有孔核壳型金纳米粒子1mL,5000r/min离心5min,弃上清液,即得,该金纳米粒子的浓度在浓缩前为0.05nmol/L左右;浓缩后为0.25nmol/L左右。
实施例2不同阴离子对于百草枯样品在1643cm-1处特征峰的信号影响
取含50μg/L百草枯的血浆溶液,加入到离心浓缩后的有孔核壳型金纳米粒子中,混匀,再加入10μL 2mol/L I-、S2-、F-、Cl-、Br-、IO3-、SO32-、SO42-、HPO42-、NO2-、CO32-、HCO3-、NO3-、CN-、SCN-溶液,混匀,采用便携式拉曼光谱仪,在激光功率150mW,积分时间10s下检测进行SERS检测信号强度。不同的阴离子对于50μg/L百草枯样品在1643cm-1处特征峰的信号影响如图3-A所示,从中可以看出:I-、S2-和F-能起到增强百草枯信号的作用,I-与S2-效果相近,F-效果相对较弱。
实施例3不同浓度KI对50μg/L血浆百草枯在1643cm-1处拉曼 信号强度的影响
取含50μg/L百草枯200μL血浆溶液,加入到离心浓缩后的有孔核壳型金纳米粒子中,混匀,再加入10μL浓度为1,5,10,20,50,100,120,150,200mmol/L的KI溶液,混匀,采用便携式拉曼光谱仪,在激光功率150mW,积分时间10s下检测进行SERS检测信号强度,每个浓度平行三次。不同浓度KI对50μg/L血浆百草枯在1643cm-1处拉曼信号强度的影响如图3-B所示。从中可以看出:对于50μg/L百草枯血浆溶液,加入10μL上述不同浓度的KI溶液,均出现百草枯的特征峰。加入100mmol/L KI溶液,百草枯的特征峰最明显。
实施例4不加KI和加入KI对50μg/L百草枯血浆样品的影响
取含50μg/L百草枯200μL血浆溶液两份,分别加入两份到离心浓缩后的有孔核壳型金纳米粒子中,混匀。取其中一份加入10μL 2mol/L的KI溶液,混匀,采用便携式拉曼光谱仪,在激光功率150mW,积分时间10s下检测进行SERS检测信号强度,另一份不加入KI溶液作对比。不加与加入0.1mol/L KI的50μg/L百草枯血浆样品的SERS谱图如图4所示,其中a为不加KI时,b为加入KI时。从图4可以看出:不加入KI溶液,百草枯的特征信号不明显,甚至有些特征峰未出现;加入KI溶液后,百草枯的拉曼特征信号明显增强。
实施例5血浆中毒样品浓度工作曲线的建立
取1mg百草枯纯品于5mL容量瓶定容得200mg/L的储备液,用超纯水稀释得到浓度为0.005,0.05,0.5,5,50mg/L的标准工作液,储存于4℃冰箱。取200μL血浆10份,分别加入一定量的百草枯标准溶液得浓度为0,0.5,1,2,5,10,20,50,100,200μg/L的一系列百草枯血浆样品。10份标准样品分别加入到10份离心浓缩后的有孔核壳型纳米金溶胶中,混匀,再分别加入10μL 2mol/L的KI溶液,混匀,采用便携式拉曼光谱仪,在激光功率150mW,积分时间10s下检测进行SERS检测信号强度。10份标准样品的SERS谱图如图5-A所示。根据1643cm-1处SERS峰强度,绘制标准曲线,如图5-B所示。从图中可以得出:对于百草枯中毒血浆样品的样品线性检测范围为1-40μg/L,线性相关系数 R2=0.983,检出限为1μg/L。
实施例6不加KI和加入KI对百草枯尿样的影响
取两份1mL空白人尿液中添加10μL浓度为5mg/L百草枯工作液,以三倍体积去离子水稀释。依次用1mL甲醇和1mL水活化Cleanert PWCX固相萃取小柱,取1ml稀释后的尿样上柱,用2mL 50%(v/v)甲醇水溶液淋洗,去除杂质,用2mL 2%(v/v)三氟乙酸水溶液洗脱待测物,获得的洗脱液,各取200μL洗脱液加入到两份离心浓缩后的有孔核壳型金纳米粒子混合,样品a加入10μL 2mol/L KI溶液混合均匀,样品b不添加KI溶液,用便携式拉曼光谱仪在激光功率150mW,积分时间10s下检测,获得的SERS谱图如图6所示。
结果显示,不加入KI溶液,百草枯的特征信号不明显,甚至有些特征峰未出现;加入KI溶液后,百草枯的拉曼特征信号明显增强。
实施例7尿液中毒样品浓度工作曲线的建立
取1mg百草枯纯品于5mL容量瓶定容得200mg/L的储备液,用超纯水稀释得到浓度为0.005,0.05,0.5,5,50mg/L的标准工作液,储存于4℃冰箱。取1mL空白人尿液9份,分别加入一定量的百草枯标准溶液得浓度为0,2,4,5,10,20,50,100,200μg/L的一系列百草枯尿液样品,以三倍体积去离子水稀释。依次用1mL分析纯甲醇和1mL水活化Cleanert PWCX固相萃取小柱,取1ml稀释后的尿样上柱,用2mL50%(v/v)甲醇水溶液淋洗杂质,用2mL 2%(v/v)三氟乙酸水溶液洗脱待测物,各取200μL洗脱液加入到9份离心浓缩后的有孔核壳型金纳米粒子混合,混匀,再分别加入10μL 2M的KI溶液,混匀,采用便携式拉曼光谱仪,在激光功率150mW,积分时间10s下检测进行SERS检测信号强度。9份标准样品的SERS谱图如图7-A所示。根据1643cm-1处SERS峰强度,绘制标准曲线,如图7-B所示。从图中可以得出结论:对于百草枯中毒尿液样品的样品线性检测范围为5-100μg/L,线性相关系数R2=0.993,检出限为4μg/L。
实施例8考察本发明的检测方法的特异性
为考察本发明的检测方法的特异性,选择与百草枯结构相似的敌草 快、4,4'-联吡啶作为对比样品,三者的化学结构如下:
a、配制浓度为50μg/L百草枯、50μg/L敌草快、500μg/L 4,4'-联吡啶混合水溶液,取200μL与离心浓缩后的有孔核壳型金纳米粒子混合,加入10μL 2mol/L KI;
b、配制浓度为50μg/L敌草快、500μg/L 4,4'-联吡啶的混合水溶液,取200μL与离心浓缩后的有孔核壳型金纳米粒子混合,加入10μL 2mol/L KI;
c、配制浓度为50μg/L百草枯、500μg/L 4,4'-联吡啶混合水溶液,取200μL与离心浓缩后的有孔核壳型金纳米粒子混合,加入10μL 2mol/L KI;
d、配制浓度为50μg/L百草枯、50μg/L敌草快、500μg/L 4,4'-联吡啶的混合水溶液,取200μL与离心浓缩后的有孔核壳型金纳米粒子混合,不加KI;
分别采用便携式拉曼光谱仪,在在激光功率150mW,积分时间10s下直接SERS检测上述a至d样品,得到SERS谱图如图8-A所示,其中:a为血浆中加入4,4'-联吡啶+百草枯+敌草快的样品加入KI时,b为血浆中加入敌草快+4,4'-联吡啶加入KI时;c为血浆中加入百草枯+4,4'-联吡啶加入KI时;d为血浆中加入4,4'-联吡啶+百草枯+敌草快不加入KI时。
制备含50μg/L百草枯、50μg/L敌草快的200μL的血浆混合样品2份,分别加入到2份离心浓缩后的有孔核壳型金纳米粒子中,混匀。取其中一份加入10μL 2mol/L的KI溶液,混匀,另一份不加入KI溶液作对比,分别采用便携式拉曼光谱仪,在激光功率150mW,积分时间10s下检测进行SERS检测信号强度,获得的SERS谱图如图8-B所示。
上述结果表明:不加入KI溶液,百草枯及敌草快的特征信号不明显;加入KI溶液后,百草枯、敌草快的拉曼特征信号增强,且各自特 征峰明显,混合不干扰,证明本方法特异性强,能实现百草枯和敌草枯中毒血浆样品的区分检测。
实施例9百草枯模拟体液(灌肠液)样品的检测
配制10μL,5mg/L的百草枯生理盐水溶液,稀释至50μg/L,作为模拟体液(灌肠液)。取稀释溶液200μL加入到1mL,经5000r/min,5min离心浓缩后的有孔核壳型金纳米粒子中,混匀,再加入10μL 2mol/L KI溶液,混匀,采用便携式拉曼光谱仪,在激光功率150mW,积分时间10s下进行SERS检测,获得的SERS谱图如图9所示。结果显示:百草枯中毒50μg/L体液样品可测定,拉曼特征峰明显。表明该方法能满足快速高灵敏检测疑似百草枯中毒患者的体液样品的诊断要求。
实施例10百草枯全血样品的检测
将4℃冷藏SD大鼠全血,放置至室温后取1mL,添加百草枯标准液得浓度为32μg/L的全血中毒样品,该样品在转速2000r/min下离心10min,取上层血浆200μL备用。将有孔核壳型金纳米粒子1mL,5000r/min离心5min,弃上清液,加入上述制备的血浆样品混匀,再加入10μL 2mol/L KI溶液,混匀,采用便携式拉曼光谱仪,在激光功率150mW,积分时间10s下检测进行SERS检测,获得的SERS谱图如图10所示。结果显示:百草枯中毒32μg/L全血样品可测定,拉曼特征峰明显。表明该方法能满足快速高灵敏检测疑似百草枯中毒患者的全血样品的诊断要求,所得结果与空白人血浆添加检测无明显区别。
实施例11百草枯中毒人员肺样品固相萃取液的检测
取-80℃,百草枯中毒患者肺组织0.5g(从医院获得),放置至室温,加入超纯水2mL,匀浆,在转速3000r/min下离心10min,取上清液,以三倍体积去离子水稀释。依次用1mL甲醇和1mL水活化Cleanert PWCX固相萃取小柱,取1ml稀释后的上清液上柱,用2mL 50%(v/v)甲醇水溶液淋洗,去除杂质,用2mL 2%(v/v)三氟乙酸水溶液洗脱待测物,获得洗脱液,取200μL洗脱液加入到离心浓缩后的有孔核壳型金纳米粒子混合。采用便携式拉曼光谱仪,在激光功率150mW,积分时间10s下进行SERS检测,获得的SERS谱图如图11所示。结果显示: 所得的百草枯中毒组织样品谱峰清晰,拉曼特征峰明显,特异性强,表明该方法能满足快速高灵敏检测百草枯中毒患者组织样品的诊断要求。
实施例12百草枯血浆样品SERS信号随时间的变化情况
取含50μg/L百草枯的血浆溶液200μL,加入到离心浓缩后的有孔核壳型金纳米粒子中,混匀,再加入10μL 2mol/L的KI溶液,混匀,采用便携式拉曼光谱仪,在激光功率150mW,积分时间10s下检测进行SERS检测信号强度。为考察方法的稳定性,采用放置的方法,分别考察放置1,2,5,10,20,40,60min重复测定,获得的SERS谱图如图12所示。结果显示:在1小时内,该方法稳定性良好,拉曼峰强度无明显变化,可满足临床诊断要求。
对于本领域的技术人员而言,本发明所述的快速检测样品中百草枯和/或敌草快的方法,完全能够临床确诊百草枯和/或敌草快中毒并简单快速地得到分析结果,也完全可以预见本发明对于中毒患者的及时救治能起到至关重要的作用。
本发明不局限于上述实施方式,对于本领域的普通技术人员来说,在不脱离本发明原理的前提下,还以做出若干改进和修饰,如,检测对象除百草枯外,其结构类似物敌草快、代谢物等荷正电分析物也可以作为本发明的检测对象。这些改进和修饰也视为本发明的保护范围之内。本说明书中未详细描述的内容属于本领域专业人员公知的现有技术。
- 还没有人留言评论。精彩留言会获得点赞!