一种松针化学成分的检测方法与流程
本发明涉及药学领域,具体涉及一种松针中化学成分的检测方法。
背景技术:
松针是松科植物马尾松pinusmassonianalamb.的针状叶,其味酸、苦、涩,性温,无毒,具有祛风燥湿,杀虫止痒,活血安神的功效[松针临床应用及药理研究概况,黄顺东,实用中医药杂志,2005,21卷第8期]。在古代我国就已利用松针来治疗疾病。据《别录》记载松针有“:主风湿疮、生毛发、安五脏”的作用《会约医镜》中记有“辟瘟疫气”明代的《本草纲目》中记载着“炙暑冻疮;去风痛脚痹,杀米虫”等等,中医认为松针味苦,无毒、性温、有祛风、活血、明目、安神、解毒、止痒、去疲劳等功效。主治流行性感冒,风湿关节痛、跌打肿痛、夜盲症、高血压、神经衰弱、冻疮、慢性支气管炎等疾病。现代研究表明松针中含有丰富的营养成分,如马尾松针中含蛋白质、脂肪、多缩戊糖、钙、磷、叶绿素、胡萝卜素、并含有18种以上的氨基酸、40余种矿物元素、多种维生素[我国松针的开发利用研究及进展,刘晓庚等,林产化工通讯,2003,第37卷第4期]。除上述营养成分外,松针中还含有多种药理活性成分,如:黄酮类化合物、莽草酸、木脂素等[松针功能性成分及应用研究进展,陈英等,安徽农业科学,2012,第40卷第10期]。
松针黄酮(pinececdieflaronoid,pf)是一种由黄酮类化合物组成的萃取物,主要是由一些黄酮和黄酮苷及其多聚合体化合物前花青素组成,研究表明pf具有具有扩张动脉血管,抑制acei酶和改善微循环,抗炎,抗氧化,消除自由基等作用[松针黄酮类物质药理作用研究进展,陈良胜等,中外医疗,2012,第17期]。刘红煜等人以高糖高脂饮食诱导肥胖大鼠造成氧化应激状态和脂代谢紊乱,以松针总黄酮灌胃给药,考察松针总黄酮抗氧化调血脂作用,结果表明,松针总黄酮能提高肝脏抗氧化酶活性,降低mda含量,可显著改善肥胖大鼠的氧化应激状态,具有调血脂作用[松针总黄酮抗氧化调血脂作用研究,刘红煜等,中医药信息,2013,第50卷第1期]。
松针作为松树类植物的主要副产物之一,是一种再生速度快、一年四季均可采收、分布广泛、天然蓄积量大、可持续利用的天然再生资源。由于松针药材来源广泛,资源丰富,且具有极好的药用价值,在《四川省藏药材标准》(2014年版)、《湖南省中药材标准》(2009年版),《北京市中药材标准》(1998年版)、《广西中药材标准》(1996年版)中均收录了该药材,但目前均无相关的含量测定项目,使得松针药材的质量控制成为一个空白。松针黄酮是松针中较为重要的一类活性成分,其临床应用已得到越来越广泛的关注,本发明人经过长期的科研实践,建立了松针中黄酮类成分的高效液相色谱检测方法,可用于松针中槲皮素、山柰酚、飞燕草素、矢车菊素等活性成分的定性和定量分析,为工业生产中松针药材的质量控制提供了可靠的支持。
技术实现要素:
本发明提供了一种松针化学成分的检测方法,采用高效液相色谱法进行检测,所述方法以十八烷基硅烷键合硅胶为色谱柱填料,以甲醇-磷酸水溶液作为流动相,梯度洗脱,其中,所述所述磷酸水溶液的体积百分比浓度为0.1%~0.5%;
本发明所述检测方法中检测波长为370nm和530nm;
优选地,所述梯度洗脱条件为:
0~20min,37%甲醇,63%磷酸水溶液;
20~25min,37%-50%甲醇,63%-50%磷酸水溶液;
25~45min,50%甲醇,50%磷酸水溶液;
优选地,所述检测方法中柱温为30℃,流速为1.0ml/min,进样量为10μl;
优选地,所述磷酸水溶液的体积百分比浓度为0.4%;
进一步,本发明所述松针化学成分检测方法包含如下所述的样品溶液制备方法:取鲜松叶,剪成小段,加入10%-30%的盐酸和甲醇混合溶液适量,回流提取,冷却,过滤,取续滤液过微孔滤膜,即得;优选地,所述样品溶液制备方法中使用30%的盐酸和甲醇混合溶液,所述回流提取时间为1h,所述盐酸和甲醇的体积比为1:4。
进一步,本发明提供一种松针化学成分的检测方法,采用高效液相色谱法进行检测,其特征在于包含以下步骤:
1)取鲜松叶3g,剪成1-2mm的小段,加入体积比为1:4的30%盐酸和甲醇混合溶液50ml,使药材被溶液覆盖,回流提取1h,冷却,过滤,取续滤液过微孔滤膜,备用;
2)将上述步骤1)中制备得到的样品溶液注入高效液相色谱仪,以十八烷基硅烷键合硅胶为色谱填料,以体积百分比浓度为0.4%的磷酸水溶液和甲醇为流动相,进行梯度洗脱,检测波长为370nm和530nm,柱温30℃,流速1.0ml/min;其中所述梯度洗脱条件为:
0~20min,37%甲醇,63%磷酸水溶液;
20~25min,37%-50%甲醇,63%-50%磷酸水溶液;
25~45min,50%甲醇,50%磷酸水溶液。
本发明的松针化学成分检测方法具有精密度高、稳定性好、重复性高的优点,该方法准确可靠,可用于松针药材的质量控制。
附图说明
图1槲皮素和山奈酚对照品色谱图
图2飞燕草素和矢车菊素对照品色谱图
图3梯度1供试品溶液色谱图(370nm)
图4梯度1供试品溶液色谱图(530nm)
图5梯度2供试品溶液色谱图(370nm)
图6梯度2供试品溶液色谱图(530nm)
图7梯度3供试品溶液色谱图(370nm)
图8梯度3供试品溶液色谱图(530nm)
图9对比例1供试品溶液色谱图(530nm)
图10对比例2供试品溶液色谱图(530nm)
图11对比例3供试品溶液色谱图(530nm)
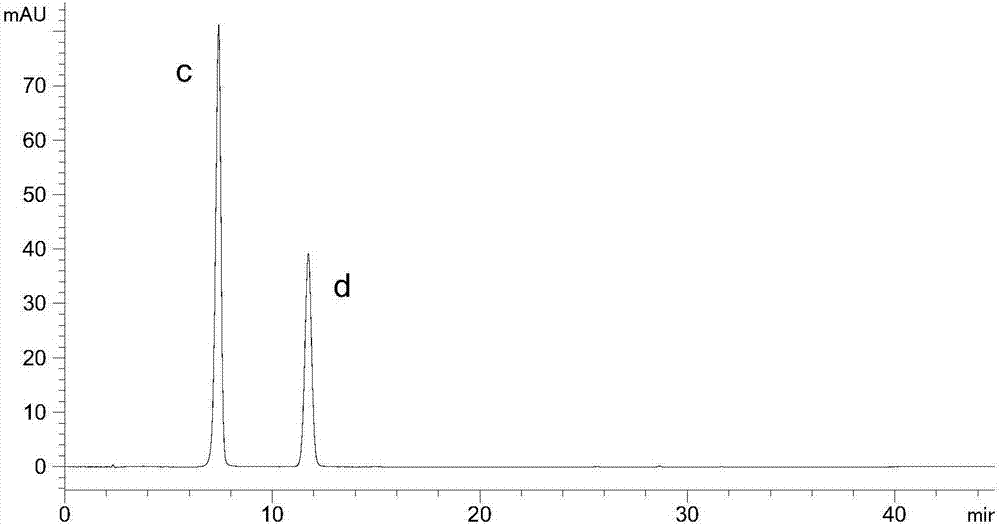
其中:a为槲皮素b为山柰酚c为飞燕草素d为矢车菊素
具体实施方式
1、实验材料
松叶为松科植物马尾松pinusmassonianalambert的针状鲜叶,由四川康弘中药材种植有限公司提供。
对照品:槲皮素,购自中国药品生物制品检定所;山柰酚,购自中国食品药品检定研究院;氯化矢车菊素,pcl-#c231,氯化飞燕草素,pcl-#de337,均购自英国purechemlandinc.(pcl)公司。
试剂:甲醇,盐酸,分析纯,成都市科龙化工试剂厂;甲醇、乙腈,色谱纯,fisher公司;磷酸,色谱纯,天津科密欧化学试剂厂。
色谱柱:agilentzorbaxsbc18(4.6×250mm,5μm,),安捷伦科技有限公司;
高效液相色谱仪:agilent1100,安捷伦科技有限公司;
0.45μm微孔滤膜,天津市津腾实验设备有限公司。
2、对照品溶液的制备
飞燕草素和矢车菊素在植物中多以糖苷形式存在,本发明中将其水解后分别检测飞燕草素和矢车菊素苷元,并以氯化飞燕草素和氯化矢车菊素为对照品,二者在液相色谱图中均为苷元色谱峰。
取槲皮素、山柰酚、氯化飞燕草素、氯化矢车菊素对照品适量,精密称定,加1%盐酸甲醇制成每1ml含槲皮素10μg、山柰酚48μg、氯化飞燕草素51μg,氯化矢车菊素30μg。
3、供试品溶液的制备
取鲜松叶3g,剪成1-2mm的小段,加入盐酸和甲醇混合溶液50ml,其中盐酸浓度在10%-30%范围内均可较好的将样品中的黄酮类化合物母核水解出来以满足定性定量检测的需求,尤以30%的盐酸效果更好,盐酸与甲醇的体积比为1:4,回流提取1h,冷却,过滤,取续滤液过0.45μm微孔滤膜,备用。
4、色谱条件
固定相:十八烷基硅烷键合硅胶流动相:为0.4%磷酸-甲醇,梯度洗脱,检测波长为370nm和530nm,柱温30℃,流速1.0ml/min。
5、洗脱条件的考察
参照上述“2”、“3”项下所述方法制备对照品和样品溶液,参照上述“4”项下色谱条件,改变不同的洗脱梯度,考察对检测结果的影响,部分实验结果如下。
梯度1:
0~20min,30%甲醇,70%磷酸水溶液;
20~25min,30%→50%甲醇,70%-50%磷酸水溶液;
25~45min,50%甲醇,50%磷酸水溶液。
检测结果:基线不平稳,矢车菊素峰附近基线升高较大,未与干扰峰完全分离,影响定量的准确性(附图3-4)。
梯度2:
0~20min,35%甲醇,65%磷酸水溶液;
20~21min,35%→50%甲醇,65%-50%磷酸水溶液;
21~41min,50%甲醇,50%磷酸水溶液。
检测结果:基线不平稳,尤其是槲皮素附近基线升高,导致无法准确定量(附图5-6)。
梯度3:
0~20min,37%甲醇,63%磷酸水溶液;
20~25min,37%→50%甲醇,63%-50%磷酸水溶液;
25~45min,50%甲醇,50%磷酸水溶液。
检测结果:目标峰实现基线分离,峰形好,基线平稳,无明显杂质干扰(附图7-8)。
根据上述实验结果可知,只有在梯度3的洗脱条件下目标成分才能实现良好的分离效果,避免杂质干扰,以实现准确的定性和定量。
6、对比实施例
对比例1
色谱柱:agilentzorbaxsbc18,流动相:甲醇-0.4%磷酸(50:50),流速1.0ml/min,检测波长370nm和530nm,柱温30℃,进样量10μl;对照品及样品溶液制备参照上述“2”、“3”项下方法。
检测结果:
飞燕草素和矢车菊素未达到基线分离,附近有其它峰干扰,且基线起伏较大,不满足分离要求,无法准确定性或定量。
对比例2
色谱柱:agilentzorbaxsbc18,流动相:乙腈-0.1%磷酸水溶液,梯度洗脱:0~15min,22%乙腈,78%磷酸溶液,15~30min,25%乙腈,75%磷酸溶液,30~45min,33%乙腈,67%磷酸溶液,流速1.0ml/min,检测波长370nm和530nm,柱温30℃,进样量10μl;对照品及样品溶液制备参照上述“2”、“3”项下方法。
检测结果:
飞燕草素和矢车菊素附近基线起伏较大,且有明显杂质干扰峰存在,分离度达不到要求,无法准确定性或定量。
对比例3
色谱柱:agilentzorbaxsbc18,流动相:0.5%磷酸-乙腈溶液,梯度洗脱(0~9min,25%→30%乙腈,75%-70%磷酸溶液;9~19min,30%乙腈,70%磷酸溶液;19~20min,30%→25%乙腈,70%-75%磷酸溶液;20~40min,25%乙腈,75%磷酸溶液),流速0.8ml.min-1,检测波长370nm和530nm,柱温30℃,进样量10μl;对照品及样品溶液制备参照上述“2”、“3”项下方法。
检测结果:
飞燕草素和矢车菊素未达到基线分离,且其中包裹其他成分,不满足定性定量要求。
7、方法学验证
1)线性回归试验
精密量取氯化飞燕草素、氯化矢车菊素、槲皮素、山柰酚对照品储备液,制得含量为每1ml含槲皮素10μg、山柰酚48μg、氯化飞燕草素51μg,氯化矢车菊素30μg,分别进样8,10,12,14,16μl测定,参照前述项“5”下梯度3进行洗脱,以峰面积为纵坐标,对照品进样量(10μg)为横坐标,进行线性回归,回归方程分别如下表1。
表1线性回归试验结果
2)氯化飞燕草素、氯化矢车菊素、槲皮素、山柰酚精密度试验
取混合对照品液,按前述项“4”下色谱条件及项“5”下梯度3进行测定,连续进样6次,测得氯化飞燕草素、氯化矢车菊素、槲皮素及山柰酚峰面积的rsd(n=6),结果如下表2。
表2精密度试验结果
3)稳定性试验
取同一供试品溶液,分别于制备后0、2、4、6、8小时,按前述项“4”下色谱条件及项“5”下梯度3进行测定,分别计算各检测指标峰面积的rsd,结果见下表3。
表3精密度试验结果
4)重复性试验
取同一批松叶,按前述项“3”下供试品溶液制备方法制备6份,按前述项“4”下色谱条件及项“5”下梯度3进行测定,以外标法计算含量,结果见下表4。
表4重复性试验结果
5)回收率试验
取已知含量的同一批的松叶药材6份含氯化飞燕草素0.084%,氯化矢车菊素0.064%,槲皮素0.029%和山柰酚含量0.15%)1.5g,精密称定,分别加入氯化飞燕草素对照品溶液,氯化矢车菊素对照品溶液,槲皮素对照品溶液和山柰酚对照品溶液适量(相当于氯化飞燕草素1.52mg,氯化矢车菊素0.91mg,槲皮素0.32mg,山柰酚1.44mg),按前述项“3”下供试品溶液制备方法制备样品溶液,按照前述项“4”下色谱条件及项“5”下梯度3进样测定,以外标法计算含量,求得氯化飞燕草素、氯化矢车菊素、槲皮素、山柰酚的加样回收率分别为106.6%、105.4%、100.1%、100.5%,rsd分别为1.6%、1.2%、0.59%、0.61%。
方法学验证试验表明,本发明检测方法精密度高、稳定性好、重复性高,该方法准确可靠,可用于松针药材的质量控制。
8、样品检测
按照上述“2”、“3”、“4”项下方法和色谱条件,及“5”项下梯度3,对3批松叶药材中的化学成分进行检测,结果如下表5:
表5样品检测结果
- 还没有人留言评论。精彩留言会获得点赞!