一种同时测定野菊花中七种成分的含量的方法与流程
本发明涉及一种同时测定野菊花中多钟成分的含量的方法,尤其是一种同时测定野菊花中七种成分的含量的方法,属于化学检测技术领域。
背景技术:
野菊花为菊科植物野菊Chrysanthemum indicum L.的干燥头状花序,具清热解毒、泻火平肝之功效,常用于疔疮痈肿,目赤肿痛,头痛眩晕。主要成分有黄酮类、萜类、挥发油、野菊花内酯、胡萝卜苷、氨基酸及微量元素等多种物质。实验研究表明野菊花具有一定的抗炎和镇痛作用,绿原酸、木犀草苷、芹菜素和蒙花苷为野菊花的抗炎药效组分。目前多采用高效液相色谱外标法对野菊花中上述成分进行含量测定,戴胜等用HPLC测定野菊花药材中8种黄酮和有机酸的含量,申海进等用RP-HPLC测定野菊花中槲皮素、木犀草素、芹菜素和刺槐素的含量。
《中国药典》2015年版野菊花控制指标为蒙花苷,单成分含量难以进行整体质量评估,但多成分质量控制要求对照品的种类比较多,而部分对照品价格昂贵甚至供应量非常少,无法满足检测需求。一测多评法(quantitative analysis of multi-components by single-marker,
QAMS)依据中药材内各有效成分间的某些内在关系,选择价廉、易得的成分为内参物,建立该成分与其他成分间的相对校准因子(relative correction factor,RCF),通过RCF计算其他成分的含量。
技术实现要素:
针对上述技术问题,本发明的目的是同时提供一种同时测定野菊花中七种成分的含量的方法,该方法高效准确、重现性良好。
为解决上述技术问题,本发明的技术方案是这样的:
一种同时测定野菊花中七种成分的含量,其特征在于,依次包括下述步骤:
1)对照品溶液的制备
分别精密称取绿原酸、咖啡酸、木犀草苷、3,5-O-二咖啡酰基奎宁酸、蒙花苷、木犀草素、芹菜素对照品适量,加甲醇溶解并定容,制成质量浓度分别为0.8469mg.mL-1、0.5670mg.mL-1、0.3967mg.mL-1、0.6738mg.mL-1、0.1962mg.mL-1、0.6244mg.mL-1、1.049mg.mL-1的对照储备液,再分别精密吸取7个对照品储备液0.6、0.9、1.3、0.8、2.5、0.8、0.5mL,置同一25mL量瓶中,加甲醇稀释至刻度,即得混合对照品溶液A,精密吸取对照品溶液A 1mL至5mL量瓶中,加甲醇至刻度,制得混合对照品溶液B,避光保存;
2)快速溶剂萃取法制备供试品溶液
取本品粉末0.25g,与1g硅藻土混合均匀填充于5mL萃取池,按快速溶剂萃取仪优化的条件进行提取,萃取溶剂为甲醇、萃取温度100℃、静态萃取时间7min、冲洗体积60%、循环萃取2次,萃取结束后,将萃取液浓缩至5mL后,转移至10mL容量瓶中,用甲醇稀释至刻度,摇匀,滤过,取续滤液,即得;
3)将对照品溶液和试品溶液分别注入色谱仪检测,色谱柱为Syncronis C18,流动相为乙腈-0.5%磷酸水溶液进行梯度洗脱;流速0.5mL.min-1,检测波长334nm,柱温35℃,进样量2μL;
4)取混合对照品溶液,分别进样0.2、0.5、1、2、3、4、5μL,在上述色谱条件下测定其峰面积,以各成分进样量为横坐标,峰面积为纵坐标,绘制标准曲线,得回归方程及相关系数。
进一步的,上述的一种同时测定野菊花中七种成分的含量,所述的梯度洗脱程序为0~4min,10%~20%乙腈,90%~80%磷酸水溶液;4~10min,20%~26%乙腈,80%~74%磷酸水溶液;10~18min,26%~30%乙腈,74%~70%磷酸水溶液;18~20min,30%~40%乙腈,70%~60%磷酸水溶液;20~20.5min,40%~80%乙腈,60%~20%磷酸水溶液;20.5~23min,80%乙腈,20%磷酸水溶液;23.1min,10%乙腈,90%磷酸水溶液。
本发明与现有技术相比,具有以下优势:
本发明提供的技术方案方法采用高效液相色谱法,以绿原酸为内参物,建立绿原酸与咖啡酸、木犀草苷、3,5-O-二咖啡酰基奎宁酸、蒙花苷、木犀草素、芹菜素的相对校正因子,并考察了相对校正因子的重现性,分别采用外标法和一测多评法进行野菊花中7种成分的含量测定。建立的校正因子重现性良好,野菊花药材采用一测多评法计算的含量值与外标法实测值之间没有明显差异,同时测定7种成分的一测多评法控制野菊花药材的质量是可行的、准确的。
附图说明
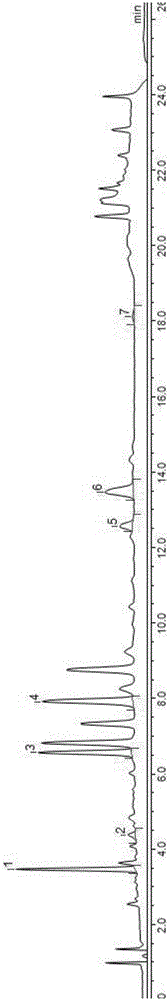
图1混合对照品的HPLC图
图2是野菊花样品的HPLC图。
具体实施方式
下面结合具体实施例对本发明的权利要求书做进一步说明,但不构成对本发明的任何限制,任何对本发明做有限次修改可得的技术方案仍然属于本发明所要保护的范围。
实施例1
1仪器与试药
1.1仪器
ThermoFisher SCITIFIC ULTIMATE 3000高效液相色谱仪(赛默飞世尔科技公司),Agilent 1290高效液相色谱仪(美国安捷伦公司),ASE350快速溶剂萃取仪(赛默飞世尔科技公司),XA205DU电子分析天平(梅特勒仪器有限公司),超纯水机(默克密理博公司)。
色谱柱:Syncronis C18(100mm×3mm,3μm,赛默飞世尔科技公司),Accucore C18(100mm×3mm,2.6μm,赛默飞世尔科技公司),BDS Hypersil C18(100mm×4.6mm,2.4μm,赛默飞世尔科技公司),Zorbax Extend C18(100mm×4.6mm,1.8μm,安捷伦科技公司),Kinetex XB-C18(100mm×4.6mm,2.6μm,广州菲罗门科学仪器有限公司),Ultracore super C18(75mm×2.1mm,2.5μm,ACE公司)。
1.2试药
对照品绿原酸(批号110753-201415,含量以96.2%计)、咖啡酸(批号110885-200102,含量以100%计)、木犀草苷(批号111720-200604,含量以100%计)、3,5-O-二咖啡酰基奎宁酸(批号111782-201405,含量以92.0%计)、蒙花苷(批号111528-200606,含量以100%计)、木犀草素(批号111520-200504,含量以92.0%计)、芹菜素(批号111901-201102,含量以99.6%计)均由中国食品药品检定研究院提供;乙腈,色谱纯,默克公司;超纯水自制;其余试剂均为分析纯。
野菊花药材均为市售品(批号1510020、1511017、150802,产地安徽;批号E5040302、1511008、1511010、1601003,产地湖北;批号151001、150806、151106,产地江西),经广西梧州食品药品检验所黄蘅副主任中药师鉴定为菊科植物野菊的头状花序。
2方法与结果
2.1色谱条件
色谱柱为Syncronis C18(100mm×3mm,3μm),流动相为乙腈-0.5%磷酸水溶液,梯度洗脱程序为0~4min,10%~20%乙腈;4~10min,20%~26%乙腈;10~18min,26%~30%乙腈;18~20min,30%~40%乙腈;20~20.5min,40%~80%乙腈;20.5~23min,80%乙腈;23.1min,10%乙腈;流速0.5mL.min-1,检测波长334nm,柱温35℃,进样量2μL。在上述色谱条件下,混合对照品和供试品中绿原酸、咖啡酸、木犀草苷、3,5-O-二咖啡酰基奎宁酸、蒙花苷、木犀草素、芹菜素分离良好,以各成分色谱峰计算的理论塔板数均不低于3000。见图1和图2,1-绿原酸,2-咖啡酸,3-木犀草苷,4-3,5-O-二咖啡酰基奎宁酸,5-蒙花苷,6-木犀草素,7-芹菜素。
2.2对照品溶液的制备
分别精密称取绿原酸、咖啡酸、木犀草苷、3,5-O-二咖啡酰基奎宁酸、蒙花苷、木犀草素、芹菜素对照品适量,加甲醇溶解并定容,制成质量浓度分别为0.8469mg.mL-1、0.5670mg.mL-1、0.3967mg.mL-1、0.6738mg.mL-1、0.1962mg.mL-1、0.6244mg.mL-1、1.049mg.mL-1的对照储备液,再分别精密吸取7个对照品储备液0.6、0.9、1.3、0.8、2.5、0.8、0.5mL,置同一25mL量瓶中,加甲醇稀释至刻度,即得混合对照品溶液A,精密吸取对照品溶液A1mL至5mL量瓶中,加甲醇至刻度,制得混合对照品溶液B(各浓度约为4μg.mL-1),避光保存。
2.3快速溶剂萃取法制备供试品溶液
取本品粉末(过三号筛)约0.25g,精密称定,与约1g硅藻土混合均匀填充于5mL萃取池,按快速溶剂萃取仪优化的条件进行提取,萃取溶剂为甲醇、萃取温度100℃、静态萃取时间7min、冲洗体积60%、循环萃取2次,萃取结束后,将萃取液浓缩至约5mL后,转移至10mL容量瓶中,用甲醇稀释至刻度,摇匀,滤过,取续滤液,即得。
2.4线性关系考察
取混合对照品溶液B,分别进样0.2、0.5、1、2、3、4、5μL,在上述色谱条件下测定其峰面积,以各成分进样量(ng)为横坐标,峰面积(Y)为纵坐标,绘制标准曲线,得回归方程及相关系数,结果表明七种成分在0.8ng~20ng范围内与峰面积呈良好线性关系,见表1。
表1野菊花中七种成分回归方程和线性范围
2.5精密度试验
取上述混合对照品溶液B,连续进样6次,记录峰面积,结果绿原酸、咖啡酸、木犀草苷、3,5-O-二咖啡酰基奎宁酸、蒙花苷、木犀草素、芹菜素峰面积的RSD依次为0.15%、0.41%、0.30%、0.23%、0.37%、0.22%、0.18%,表明进样精密度良好。
2.6重复性试验
取野菊花样品(批号151001)6份,按“2.3”项下方法制备供试品溶液,按上述色谱条件进行测定,计算绿原酸、咖啡酸、木犀草苷、3,5-O-二咖啡酰基奎宁酸、蒙花苷、木犀草素、芹菜素含量分别为1.509mg.g-1、0.701mg.g-1、1.986mg.g-1、2.491mg.g-1、0.394mg.g-1、0.854mg.g-1、1.289mg.g-1,RSD依次为0.72%、1.0%、0.87%、0.51%、1.3%、0.66%、1.0%,表明方法重复性良好。
2.7稳定性试验
取同一供试品(批号151001),按“2.3”项下方法制备供试品溶液,室温下放置,分别在0、4、8、12、16、20、24h测定,记录峰面积,结果绿原酸、咖啡酸、木犀草苷、3,5-O-二咖啡酰基奎宁酸、蒙花苷、木犀草素、芹菜素峰面积的RSD依次为0.38%、0.92%、0.43%、0.57%、0.27%、0.46%、0.96%,表明供试品溶液在24h内稳定性良好。
2.8加标回收率试验
取已测定含量的野菊花样品(批号151001)9份,编号1~9号,每份0.25g,精密称定,精密移取上述7种对照储备液各0.2mL、0.4mL、0.6mL三个水平,挥干溶剂后分别定量转移至1~3号、4~6号、7~9号样品池中,按“2.3”项下方法制备供试品溶液,在上述色谱条件进行测定,分别计算绿原酸、咖啡酸、木犀草苷、3,5-O-二咖啡酰基奎宁酸、蒙花苷、木犀草素、芹菜素的平均回收率,结果详见表2,各成分平均回收率为98.46%~103.67%,RSD为1.43%~2.67%,表明该方法的准确度良好。
表2七种成分加样回收率试验结果
2.9一测多评(QAMS)法
2.9.1相对校正因子的确定
取“线性关系考察”项下结果,以绿原酸为内参物(S),按公式fsi=fs/fi=(As×Ci)/(Ai×Cs)(As、Cs分别为内参物对照品的峰面积和浓度,Ai、Ci分别为待测成分对照品的峰面积和浓度)计算咖啡酸(A)、木犀草苷(B)、3,5-O-二咖啡酰基奎宁酸(C)、蒙花苷(D)、木犀草素(E)、芹菜素(F)的相对校正因子fS/A、fS/B、fS/C、fS/D、fS/E、fS/F,结果见表3。
表3以绿原酸为内参物的fS/i测定结果
2.9.2相对校正因子重现性考察
2.9.2.1不同仪器对f的影响
试验考察了两种高效液相色谱系统:THERMO SCITIFIC ULTIMATE 3000高效液相色谱仪(美国赛默飞世尔科技公司)和Agilent 1290高效液相色谱仪(美国安捷伦公司)对各成分f的影响,结果RSD依次为1.32%、1.61%、1.27%、1.51%、2.32%、1.33%,表明不同仪器对各成分的f无显著影响。
2.9.2.2不同色谱柱对f的影响
试验考察了6种色谱柱Syncronis C18、Accucore C18、BDS Hypersil C18、Zorbax Extend C18、Kinetex XB-C18、Ultracore super C18对各成分f的影响,结果RSD依次为2.57%、2.16%、1.92%、0.74%、2.31%、1.24%,表明此6根色谱柱对各成分的f无显著影响。
2.9.2.3不同柱温对f的影响
试验考察了不同柱温(25、30、35、40℃)对各成分f的影响,结果RSD依次为1.80%、3.67%、3.39%、2.01%、1.57%、1.90%,表明柱温对各成分的f无显著影响。
2.9.2.4不同实验人员操作对f的影响
考察了3名实验人员对各f的影响,结果RSD依次为1.79%、3.46%、3.48%、2.03%、1.59%、1.92%,表明不同操作人员所得各成分的f无显著差别。
2.9.2.5f重现性影响因素综合
综合以上各种因素,咖啡酸、木犀草苷、3,5-O-二咖啡酰基奎宁酸、蒙花苷、木犀草素、芹菜素f的RSD依次为2.04%、3.70%、2.21%、1.36%、2.22%、1.37%。结果见表4。
表4各考察因素对fS/i的影响
2.9.3色谱峰定位参数考察
利用相对保留时间(ri/s)进行峰定位,ri/s=tR(i)/tR(s)(i为待测成分,s为内参物),测得咖啡酸、木犀草苷、3,5-O-二咖啡酰基奎宁酸、蒙花苷、木犀草素、芹菜素和绿原酸的ri/s结果,见表5。不同色谱条件所得到相对保留值变动较小,RSD≤5%,表明采用ri/s进行峰的定位较为可行。
表5不同仪器和色谱柱测得的ri/s
2.9.4 QAMS法与外标法测定结果的比较
取10批野菊花样品,每批3份,按“2.3”项下方法制备供试品溶液,按上述色谱条件,分别精密吸取混合对照品溶液B和供试品溶液各2μL进行测定,记录峰面积,分别用外标法与一测多评法计算其含量并对结果进行比较,测定结果见表6,2种方法所得值的偏差均在3%以内,由此说明一测多评法用于野菊花药材的多成分质量评价研究是可行的。
表6 QAMS法与外标法测定结果(mg.g-1)
3讨论
供试品溶液制备方法的选择快速溶剂萃取是在高温和高压下用溶剂对固体或半固体样品进行萃取的新颖前处理方法,本试验采用快速溶剂萃取法制备供试品溶液,并利用正交实验优化了影响快速溶剂萃取的主要因素,最终确定萃取条件为:萃取溶剂甲醇、萃取温度100℃、静态萃取时间7min、冲洗体积60%、循环萃取2次。
QAMS法对待测色谱峰定位重现性本试验通过考察不同品牌仪器和色谱柱中各待测成分与绿原酸内参物相对保留时间的重现性,发现可通过绿原酸与野菊花中另外6种成分的相对保留时间实现色谱峰定位。
相对校正因子重现性本试验选用价廉易购的绿原酸为内参物,建立该成分与其余6种野菊花成分间的相对校正因子。通过对10批不同产地的野菊花中药材的测定,验证了该分析方法的线性、精密度、重现性、稳定性以及QAMS法的准确性,并对不同品牌仪器、色谱柱、柱温、实验人员等影响因素考察QAMS法的重现性,结果表明采用QAMS法计算的结果与外标法测定结果无显著性差异,可以在缺少咖啡酸、木犀草苷、3,5-O-二咖啡酰基奎宁酸、蒙花苷、木犀草素、芹菜素对照品的情况下,通过相对校正因子计算其量。实验建立的QAMS法为野菊花药材的质量控制提供了新的方法。
以上所述的仅为本发明的较佳实施例,凡在本发明的精神和原则范围内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!