厚藤HPLC指纹图谱和UPLC指纹图谱的建立方法与流程
本发明属于中药指纹图谱技术领域,尤其涉及厚藤hplc指纹图谱和uplc指纹图谱的建立方法。
背景技术:
中药指纹图谱概念的起源是一种刑侦手段——“指纹鉴定”。中药指纹图谱技术分为中药生物指纹图谱和中药化学指纹图谱,其中,中药化学指纹图谱应用了非同种植物化学特征的多样性及同种植物化学成分的相似性原理,中药化学成分复杂多变,受自然环境、生长年限、采收炮制方法的影响变化较大,然而同种植物受制于基因调控,次生代谢的主成分具有稳定性和唯一性,同理不同种植物间的主成分类别有显著差异性。利用该原理,借助现代色谱技术,不但可以鉴别中药真伪、质量优劣、品质均一性,而且现代分析仪器可反馈出整体、大数据量信息,包括已知成分、未知成分;主要成分、微量成分;药效成分、当前无药效研究成分等综合信息。所以,中药指纹图谱技术可用于有效成分不明确的中药质量控制,也可为中药产区、采收、加工、储存期限等提供控制。但将中药指纹图谱应用到实际生产加工时,必须建立计算机模式识别系统,而化学计量学作为桥梁至关重要。化学计量学研究是一门涉及化学、统计学及计算机科学的交叉学科,其最大优点就是可对未知的、无规律的化学研究数据进行多变量分析,挖掘到更多隐藏的信息,对于多组分、多靶点的中药具有重大意义。
中药指纹图谱技术近年来发展迅速,包括薄层扫描(tlcs)、高效液相色谱法(hplc)、气相色谱法(gc)和高效毛细管电泳法(hpce)等色谱法以及紫外光谱法(uv)、红外光谱法(ir)、质谱法(ms)、核磁共振法(nmr)和x-射线衍射法等光谱法。目前,以色谱方法最为常用,由于hplc具有分离效能高、选择性高、检测灵敏度高、分析速度快、应用范围广等特点,已成为公认的一种常规分析手段。
超高效液相色谱(ultraperformanceliquidchromatography,uplc)是在传统的高效液相色谱(hplc)基础上发展起来的,和hplc相比,具有更高的分离度、速度和灵敏度等优点。目前多应用于代谢组学分析及其他一些生化领域,在天然产物的分析方面运用也逐渐兴起。使用uplc进行指纹图谱研究,进而建立新型质量控制方法,对于中药质量控制研究领域的发展将是一个极大的促进。
目前,关于厚藤的中药指纹图谱鲜有报道。
技术实现要素:
本发明要解决的技术问题是提供厚藤hplc指纹图谱和uplc指纹图谱的建立方法,为厚藤药材的质量控制提供更为科学、合理、详实的依据。
为解决上述技术问题,本发明以下技术方案:
厚藤hplc指纹图谱的建立方法,包括以下步骤:
(1)制备对照品溶液:以甲醇为溶剂配制成含绿原酸0.5998mɡ/ml、咖啡酸0.0503mɡ/ml、异槲皮苷0.0698mɡ/ml、异绿原酸b0.4001mɡ/ml、异绿原酸a0.8001mɡ/ml、异绿原酸c0.6005mɡ/ml的混合对照品溶液;
(2)供试品溶液的制备:精密称定厚藤药材粉末约2ɡ,置具塞锥形瓶中,加入70%甲醇20ml,称定重量,超声1h,取续滤液2ml过0.22μm微孔滤膜,备用;
(3)色谱条件:色谱柱为安捷伦色谱柱,采用梯度洗脱,流动相为乙腈-0.1%磷酸水系统,紫外检测波长为254nm;
(4)测定:精密吸取供试品溶液注入高效液相色谱仪测定,得到hplc指纹图谱。
步骤(1)按以下操作进行:精密称定适量绿原酸、咖啡酸、异槲皮苷、异绿原酸b、异绿原酸a、异绿原酸c对照品,加甲醇分别定容于5ml棕色容量瓶,配制成绿原酸3.0080mɡ/ml、咖啡酸2.0960mɡ/ml、异槲皮苷2.0300mɡ/ml、异绿原酸b3.0400mɡ/ml、异绿原酸a3.0400mɡ/ml、异绿原酸c3.0480mɡ/ml的标准溶液;精密移取以上标准溶液配制成含绿原酸0.5998mɡ/ml、咖啡酸0.0503mɡ/ml、异槲皮苷0.0698mɡ/ml、异绿原酸b0.4001mɡ/ml、异绿原酸a0.8001mɡ/ml、异绿原酸c0.6005mɡ/ml的混合对照品溶液。
步骤(3)的色谱条件为:色谱柱:anglienthc-c18(2)色谱柱,250mm×4.6mm,5μm;流动相:乙腈-0.1%磷酸水溶液;流速:1.0ml/min;柱温:30±5℃;检测波长:λ=254nm;进样量:5μl;洗脱梯度,a为乙腈:0~10min,7%~8%a;10~20min,8%~12%a;20~30min,12%~16%a;30~40min,16%~17%a;40~50min,17%~19%a;50~65min,19%~20%a;65~70min,25%~27%a;70~75min,25~27%a;85~86min,36~40%a;86~92min,40%~40%a;92~97min,40%~85%a。
厚藤uplc指纹图谱的建立方法,包括以下步骤:
(1)制备对照品溶液:以甲醇为溶剂配制成含绿原酸0.5998mɡ/ml、咖啡酸0.0503mɡ/ml、异槲皮苷0.0698mɡ/ml、异绿原酸b0.4001mɡ/ml、异绿原酸a0.8001mɡ/ml、异绿原酸c0.6005mɡ/ml的混合对照品溶液;
(2)供试品溶液的制备:精密称定厚藤药材粉末约2ɡ,置具塞锥形瓶中,加入70%甲醇20ml,称定重量,超声1h,取续滤液2ml过0.22μm微孔滤膜,备用;
(3)色谱条件:色谱柱为c18色谱柱,采用梯度洗脱,流动相为乙腈-0.2%磷酸水溶液,紫外检测波长为254nm;
(4)测定:精密吸取供试品溶液注入超高效液相色谱仪测定,得到uplc指纹图谱。
步骤(1)按以下操作进行:精密称定适量绿原酸、咖啡酸、异槲皮苷、异绿原酸b、异绿原酸a、异绿原酸c对照品,加甲醇分别定容于5ml棕色容量瓶,配制成绿原酸3.0080mɡ/ml、咖啡酸2.0960mɡ/ml、异槲皮苷2.0300mɡ/ml、异绿原酸b3.0400mɡ/ml、异绿原酸a3.0400mɡ/ml、异绿原酸c3.0480mɡ/ml的标准溶液;精密移取以上标准溶液配制成含绿原酸0.5998mɡ/ml、咖啡酸0.0503mɡ/ml、异槲皮苷0.0698mɡ/ml、异绿原酸b0.4001mɡ/ml、异绿原酸a0.8001mɡ/ml、异绿原酸c0.6005mɡ/ml的混合对照品溶液。
步骤(3)的色谱条件为:色谱柱:acquityuplcbehc18色谱柱,型号:100mmx2.1mm,1.7μm;流动相:乙腈-0.2%磷酸水溶液;洗脱程序,a为乙腈:0~3min,13%~13%a;3~4min,13%~19%a;4~8min,19%~19%a;8~9min,19%~25%a;9~12min,25%~35%a;12~14min,35%~95%a;流速:0.2ml/min;柱温:35±5℃;检测波长:λ=254nm;进样量:2μl。
hplc指纹图谱共有峰13个,其中六个峰分别为绿原酸、咖啡酸、异槲皮苷、异绿原酸b、异绿原酸a、异绿原酸c的色谱峰。
uplc指纹图谱共有峰16个,其中六个峰分别为绿原酸、咖啡酸、异槲皮苷、异绿原酸b、异绿原酸a、异绿原酸c的色谱峰。
为提高厚藤药材的质量控制和真伪鉴别的水平,发明人建立了厚藤hplc/uplc指纹图谱的建立方法,采用高效/超高效液相色谱,以厚藤中的主要成分绿原酸、咖啡酸、异槲皮苷、异绿原酸b、异绿原酸a、异绿原酸c为参照物,据此获得了厚藤药材共有模式的hplc/uplc指纹图谱。其中,hplc指纹图谱中可以检测到13个共有峰,uplc指纹图谱可以检测到16个共有峰,10批厚藤药材hplc指纹图谱与共有模式的相似度均大于0.956,10批厚藤药材uplc指纹图谱与共有模式的相似度均大于0.945,厚藤hplc指纹图谱、uplc指纹图谱及其共有模式均可为厚藤的真伪鉴别、质量优劣提供科学的依据。其中,厚藤的uplc指纹图谱可提供更多色谱峰信息,聚类分析及主成分分析的分类结果较hplc指纹图谱更详细。在现有仪器设备条件下,hplc厚藤指纹图技术及其共有模式更具有普适性,uplc指纹图谱及其共有模式更适用于批次数量多、单批次样品量不足、成分复杂的样品分析,总之,本发明的hplc和uplc指纹图谱都可为厚藤药材的质量控制提供更为科学、合理、详实的依据,可根据实际情况运用。
附图说明
图1是乙腈-0.1%磷酸为流动相的厚藤药材hplc色谱图。
图2是254nm下厚藤的hplc色谱图。
图3是使用安捷伦色谱柱的hplc色谱图。
图4是梯度2洗脱出液相色谱图。
图5是以70%甲醇为提取溶剂的样品hplc色谱图。
图6是6种混合对照品的hplc,图中:峰s1:绿原酸峰s2:咖啡酸峰s3:异槲皮苷;峰s4:异绿原酸b峰s5:异绿原酸a峰s6:异绿原酸c。
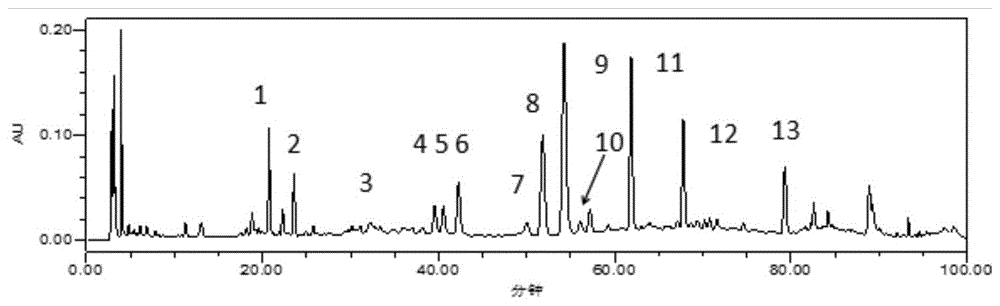
图7是厚藤hplc色谱图,图中:峰1:绿原酸峰2:咖啡酸峰6:异槲皮苷;峰8:异绿原酸b峰9:异绿原酸a峰11:异绿原酸c。
图8是10批厚藤药材hplc色谱图叠加图。
图9是10批厚藤hplc色谱图共有模式r。
图10是不同产地厚藤的聚类分析图。
图11是各个批次药材的主成分得分图
图12是ⅰ类厚藤液相色谱图的共有模式r1液相色谱图。
图13是ⅱ类厚藤液相色谱图的共有模式r2液相色谱图。
图14是以乙腈-0.2%磷酸为流动相的uplc色谱图。
图15是洗脱程序2下的uplc色谱图。
图16是柱温为35℃时的uplc色谱图。
图17是流速为0.2ml/min的uplc色谱图。
图18是六种混合标准品的uplc色谱图。
图19是厚藤uplc色谱图。
图20是10批厚藤药材的uplc色谱图叠加图。
图21是10批厚藤药材的uplc色谱图的共有模式r’。
图22是不同产地厚藤的聚类分析图。
图23是各个批次药材的pca1,pca2,pca3主成分的得分图。
图24是各个批次药材的pca1,pca2,pca4主成分的得分图。
图25是ⅰ类厚藤的uplc色谱图的共有模式r’1。
图26是ⅱ类厚藤的uplc色谱图的共有模式r’2。
图27是iii类厚藤的uplc色谱图的共有模式r’3。
具体实施方式
一、厚藤药材的hplc指纹图谱研究
1.试验仪器与材料试剂
1.1试验仪器
watershplc高效液相色谱仪,包括四元超高压溶剂系统、自动进样样本管理器、pda检测器、empower2色谱工作站(waters公司);十万分之一电子分析天平(赛多利斯科学仪器北京有限公司,型号:sqp);超声波清洗机(宁波新芝生物科技股份有限公司,型号:sb25-12d);电热恒温水浴锅(北京市永光明医疗仪器有限公司,型号:dzkw-d-6);万能粉碎机(天津太斯特仪器有限公司,型号:wnd-200);电热恒温鼓风干燥箱(上海精宏试验设备有限公司,型号:dhg-9240a)。
1.2药品与试剂
药品:绿原酸(批号:20170903)、咖啡酸(批号:20171224)、异槲皮苷(批号:20180105)、异绿原酸b(批号:20171029)、异绿原酸a(批号:20170913)、异绿原酸c(批号:20171106),含量≧98%,均购自于上海融禾医药科技发展有限公司
试剂:乙腈、甲醇(色谱纯,美国飞世尔试剂公司);甲醇(分析纯,成都科隆化学品有限公司,批号:2017030201);95%乙醇(分析纯,成都科隆化学品有限公司,批号:2017031001);磷酸(分析纯,成都金山化学试剂有限公司,批号:20151006);水为超纯水
1.3药材
厚藤采集于广西及海南,总计20批,经广西中医药大学李永华教授鉴定为旋花科植物厚藤ipomoeapes-caprae(l.)sweet的干燥茎叶,新鲜药材置烘箱中55℃烘干,粉碎,药材粉末过55目筛备用,厚藤药材样品来源信息如表1和表2所示:
表1不同产地厚藤的样品来源信息
表2不同采收期厚藤的样品来源信息
2方法与结果
2.1色谱条件
色谱柱:anglienthc-c18(2)色谱柱(250mm×4.6mm,5μm);流动相:乙腈-0.1%磷酸水溶液;流速:1.0ml/min;柱温:30±5℃;检测波长:λ=254nm;进样量:5μl;洗脱梯度(即梯度2,a为乙腈):0~10min,7%~8%a;10~20min,8%~12%a;20~30min,12%~16%a;30~40min,16%~17%a;40~50min,17%~19%a;50~65min,19%~20%a;65~70min,25%~27%a;70~75min,25~27%a;85~86min,36~40%a;86~92min,40%~40%a;92~97min,40%~85%a。
2.2供试品的制备
加热回流提取:精密称定厚藤药材粉末(晒干粉碎且过50目筛)约2ɡ,置具塞锥形瓶中,加入一定浓度溶剂20ml,称定重量并记录,85℃加热回流提取1h,待提取结束,取出锥形瓶放冷并擦干外壁,称定重量并记录,补足重量,摇匀过滤,取续滤液2ml过0.22μm微孔滤膜,转移至进样瓶备用。
超声提取:精密称定厚藤药材粉末(过50目筛)约2ɡ,置具塞锥形瓶中,加入一定浓度溶剂20ml,称定重量,超声1h,放冷并擦干外壁,补足重量,摇匀过滤。取续滤液2ml过0.22μm微孔滤膜,转移至进样瓶备用。
2.3对照品溶液的制备
精密称定适量绿原酸、咖啡酸、异槲皮苷、异绿原酸b、异绿原酸a、异绿原酸c等对照品,加甲醇分别定容于5ml棕色容量瓶,配制成绿原酸3.0080mɡ/ml、咖啡酸2.0960mɡ/ml、异槲皮苷2.0300mɡ/ml、异绿原酸b3.0400mɡ/ml、异绿原酸a3.0400mɡ/ml、异绿原酸c3.0480mɡ/ml的标准溶液。精密移取以上标准溶液配制成含绿原酸0.5998mɡ/ml、咖啡酸0.0503mɡ/ml、异槲皮苷0.0698mɡ/ml、异绿原酸b0.4001mɡ/ml、异绿原酸a0.8001mɡ/ml、异绿原酸c0.6005mɡ/ml的混合对照品溶液。
2.4hplc色谱条件的建立
2.4.1流动相的选择
本试验试用了甲醇-水、甲醇-0.1%磷酸、乙腈-0.1%磷酸、乙腈-0.12%磷酸等四个流动相系统。使用甲醇-水系统,色谱图分离效果差,推测可能多数成分具有羟基、羧基、苯酚等显酸性官能团,流动相系统更换为甲醇-0.1%磷酸水系统,对0~90min色谱峰分离度有很大提高,但是在90~100min有弱极性成分产生的较大包峰。为分离包峰中弱极性成分,流动相系统更换为极性较弱的乙腈-0.1%磷酸水系统,30~50min及90~100min色谱峰得到很好的分离,结果如图1所示,以乙腈-0.1%磷酸水系统所得色谱峰数量最多、分离度最好、基线较平。因此,后续试验以乙腈-0.1%磷酸水系统作为厚藤hplc指纹图谱研究的流动相。
2.4.2波长的选择
本试验对厚藤药材样品的hplc全谱进行全波长扫描,选择对大部分物质都有紫外吸收较多的波长254nm、280nm、300nm、330nm、365nm等,结果表明,254nm下,5~25min的色谱峰数量较其他波长下多,各个批次厚藤的色谱峰较其他波长下的峰面积大、分离度好。因此,取254nm作为厚藤指纹图谱的紫外检测波长。
2.4.3同一规格不同品牌色谱柱的选择
本试验选择月旭、菲罗门、安捷伦等不同品牌同一规格(250mm×4.6mm,5μm)的色谱柱,试验结果显示,40~50min、80~90min时间段内,安捷伦色谱柱较其他品牌色谱柱出峰数量多、色谱峰峰面积更大、分离度好。所以,选择安捷伦色谱柱作为后续试验用色谱柱。
2.4.4洗脱程序的选择
本试验优选出三个不同的洗脱梯度进行比较。结果显示,其中梯度2洗脱出的hplc色谱峰在20~30min、60~70min色谱峰分离度较好、基线平滑、无包峰,所以,选择梯度2作为后续试验用洗脱程序。
2.4.5空白溶剂试验
按照在“2.1”项下色谱条件使用空白溶剂进样,结果表明,在该条件下,紫外灯对样品溶剂基本无吸收,溶剂对色谱图不产生影响。
2.5样品制备方法的优化
2.5.1提取方法的选择
分别采用超声、加热回流法提取两份样品,按照“2.1”项下色谱条件进样,结果显示,不同提取方法对色谱峰数量基本无影响,且超声提取操作简单、快捷、节约能源。因此,后续试验选择超声提取法提取样品。
2.5.2提取溶剂及浓度的选择
现代化学研究表明厚藤中含多种极性物质,本试验使用甲醇以及乙醇作为提取溶剂,考察20%、50%、70%、95%等不同浓度甲醇及乙醇对厚藤hplc指纹图谱的影响。采用“2.2”项下方法提取8份样品,按照“2.1”项下色谱条件进样,结果显示,0~15min,以甲醇为提取溶剂的色谱图峰数量较乙醇多;90-100min,以70%甲醇为提取溶剂的色谱图色谱峰数量较多。因此,后续试验以70%甲醇为提取溶剂。
2.6方法学考察
2.6.1精密度试验
精密称取厚藤药材粉末,照“2.2”项下方法制备供试品溶液,按照“2.1”项下色谱条件连续进样6次,检测指纹图谱,选取13个分离度>1.5、峰面积较大的色谱峰计算相对峰面积,采用国家药典委员会组织开发的《中药色谱指纹图谱相似度评价系统》(v2.0)计算相似度,结果如表3~5所示,同一编号色谱峰在不同进样时间的相对峰面积的相对标准偏差rsd<3.10%,相对保留时间rsd<0.20%,六次进样的色谱峰与共有模式r的相似度均为1.000,说明仪器的精密度合格,符合指纹图谱测定要求。
表3精密度试验-相对峰面积(n=6)
表4精密度试验-相对保留时间(n=6)
表5精密度试验相似性结果
2.6.2重复性试验
精密称取同一批样品各6份,分别制备供试品溶液,依次进样,选取13个分离度>1.5、峰面积较大的色谱峰计算相对峰面积,采用国家药典委员会组织开发的《中药色谱指纹图谱相似度评价系统》(v2.0)计算相似度,结果显示,各色谱峰相对峰面积的相对标准偏差rsd<3.10%,相对保留时间rsd<0.30%,六次进样的色谱峰与共有模式r的相似度均为1.000,表明该方法的重复性好,符合hplc指纹图谱测定的要求。
2.6.3稳定性试验
取同一供试品溶液,分别在0h,2h,4h,8h,i2h,24h时进样5ul,检测指纹图谱,选取13个分离度>1.5、峰面积较大的色谱峰计算相对峰面积,采用国家药典委员会组织开发的《中药色谱指纹图谱相似度评价系统》(v2.0)计算相似度,结果显示,各色谱峰相对峰面积的相对标准偏差rsd<3.00%,相对保留时间rsd<0.40%,各样品色谱图与共有模式r的相似度均大于0.987,说明样品在该方法下,24h内成分稳定,符合hplc指纹图谱测定的要求。
2.7结合化学计量学建立厚藤的hplc指纹图谱
2.7.1色谱峰的指认
按“2.2”项下方法制备样品,取“2.3”项下混合对照品溶液,采用“2.1”项下色谱条件进样,在同一洗脱程序下依次进样。根据相对保留时间tr,色谱峰光谱图一致性原则,从13个共有峰中指认六个峰,即编号峰1、2、6、8、9、11分别为绿原酸、咖啡酸、异槲皮苷、异绿原酸b、异绿原酸a、异绿原酸c,结果如图6~7所示。
2.7.2参照峰的确定
本试验选择分离度较好、峰面积较大、保留时间适宜的9号色谱峰(异绿原酸a)作为参照峰s。
2.7.3共有指纹峰的指定及相似度评价
10批厚藤药材共有峰编号如图1,本试验采用国家药典委员会组织开发的《中药色谱指纹图谱相似度评价系统》(v2.0)评价10批厚藤药材的色谱图相似度,10批厚藤药材与共有模式r的相似度值范围在0.956~1.000之间,建立多个批次下的色谱叠加图,生成共有模式r,如图8~9。
表610批厚藤药材13个共有峰的相对峰面积
表710批厚藤药材的相似度结果
2.7.4厚藤指纹图谱的聚类分析(hca)
聚类分析(hierarchicalclusteringanalysis)是常用的探索性统计方法,适用于分析受多因素影响的样本,在中药真伪鉴别、质量评价、品种分类等方面广泛应用。本试验将10批不同产地厚藤药材的13个共有峰的相对峰面积作为变量,得到10×13阶原始数据矩阵,导入spss19.0软件,采用离差平方和法(ward法),选用欧氏距离(euclidean)为测度,对数据进行标准化转换后进行q型聚类分析以挖掘厚藤指纹图谱数据,得到厚藤药材的聚类分析谱图,如图10,聚类分析将10批厚藤分为了2类,ⅰ类包括第2,6,9批,ⅱ类包括第1,3,4,5,7,8,10批次。
2.7.5厚藤指纹图谱的主成分分析(pca)
主成分分析法(principalcomponentsanalysis)是将多个变量转化为少数的独立综合变量的降维分类分析法。主成分个数的选择原则包括:保留累计贡献率大于70%的主成分;保留特征值大于1的主成分。本试验将10批不同产地厚藤药材的13个共有峰的相对峰面积作为变量,对数据降维后进行因子分析。
表8主成分分析解释总变量
结果如表8所示,前三个主成分的特征值均大于1,第一主成分方差贡献率为47.464%,第二主成分方差贡献率为20.664%,第三主成分方差贡献率为13.324%,pca1、pca2、pca3的累计贡献率为81.45%,可表达厚藤药材的大部分信息。
表9旋转成份矩阵
第一主成分的特征值最大,可较为全面得描述指纹图谱的信息,根据主成分分析的成分旋转成分矩阵表9,其中色谱峰峰号1、4、8、11与第一主成分的相关性系数分别为0.949、0.679、0.907、0.942,表明4个峰直接影响厚藤药材的分类、品质优劣,其中峰号1、8、11已指认为绿原酸、异绿原酸b、异绿原酸c,峰号4色谱峰有待进一步研究指认。
主成分得分图见图11,10批厚藤药材分为ⅰ类、ⅱ类,所得到的分类结果与聚类分析一致,根据分类结果,采用相似度软件生成ⅰ类、ⅱ类厚藤液相色谱图的共有模式,见图12~13,该共有模式可分别为ⅰ类、ⅱ类厚藤的质量控制提供科学依据。
3.讨论与小结
本试验在方法学考察中引入了相似度分析,不再局限于将有限的共有峰作为方法适用性的考察对象,相似度分析结果反映了不同批次样品图谱与共有模式间的整体信息匹配度,使方法学考察的结果更全面、可靠、科学。
本试验建立了10批不同产地厚藤的hplc色谱图,指认了峰1、2、6、8、9、11分别为绿原酸、咖啡酸、异槲皮苷、异绿原酸b、异绿原酸a、异绿原酸c,该图谱相对保留时间的相对标准偏差在合理范围内,各批次药材的图谱与共有模式的相似度均大于0.956,表明该共有模式较好地反映了各批次厚藤的整体信息,6个归属峰体现了厚藤药材的专属性,该共有模式及6个归属峰的指认均可为厚藤药材的真伪鉴别、质量评价提供科学依据。
本试验对10批厚藤药材的13个共有峰进行聚类分析及主成分分析,色谱峰1、4、8、11是直接影响厚藤药材的分类、品质优劣的关键色谱峰。根据聚类、相似度分析结果,ⅰ类药材分别产自广西防城港市江山半岛(三块石)、北仑河口红树林自然保护区、钦州市三娘湾,ⅱ类药材分别产自广西城港市防城区、城港市江山半岛(脯鱼湾)、城港市江山半岛(白浪滩)、城港市江山半岛(白沙湾)、广西北海市合浦县白沙镇独山村、海南陵水黎族自治县陵水英州镇清水湾、海南海口市秀英区。根据以上结果,药材分类与产地无显著关联,推测可能是由于样本的采收季节不同,造成了同一产地间有较大的差异,但根据相似性结果,产自海南的ip-8、ip-10与共有模式r的相似度分别为0.984、0.982,两者较为接近,相似度值受采收季节影响较小。
采用相似度软件,生成10批次药材图谱的共有模式及ⅰ类、ⅱ类厚藤液相色谱图的共有模式,第2、6、9批与10批次药材共有模式r的相似度均大于0.957,第1、3、4、5、7、8、10批次与10批次药材共有模式图谱r的相似度均大于0.956,ⅰ类药材(即第2,6,9批)与其相应共有模式图谱r1的相似度均大于0.963,ⅱ类药材(即第1,3,4,5,7,8,10批次)与其相应共有模式r2的相似度均大于0.971。根据以上结果可知,共有模式r1、r2与厚藤药材的相似度较10批次药材共有模式图谱r更高,可更好地反映各批次厚藤的整体信息。
二、厚藤uplc指纹图谱研究
1.试验材料、仪器与试剂
1.1试验仪器
watersacquityuplc超高效液相色谱仪,包括四元超高压溶剂系统、白动进样恒温样本管理器、pda测器、empower2色谱工作站(waters公司);watersacquityuplcbehc18色谱柱(waters公司,型号:100mmx2.1mm,1.7μm);万分之一电子分析天平(德国赛多利斯公司,型号:sqp);超声波清洗器(昆山市超声仪器有限公司,型号:kq5200b)
1.2材料与试剂同一“1.2”项下材料试剂
1.3药材同一“1.3”项下表1
2.试验与结果
2.1色谱条件
色谱柱:acquityuplcbehc18色谱柱(waters公司,型号:100mmx2.1mm,1.7μm);流动相:乙腈(a)-0.2%磷酸水溶液(b);洗脱程序(即梯度2):0~3min,13%~13%a;3~4min,13%~19%a;4~8min,19%~19%a;8~9min,19%~25%a;9~12min,25%~35%a;12~14min,35%~95%a;流速:0.2ml/min;柱温:35±5℃;检测波长:λ=254nm;进样量:2μl。
2.2供试品的制备
精密称定厚藤药材粉末(过50目筛)约0.2ɡ,置塞锥形瓶中,加入70%甲醇20ml,称定重量,超声1h,放冷,擦干外壁,补足重量,摇匀过滤。取续滤液2ml过0.22μm微孔滤膜,转移至进样瓶备用。
2.3对照品的制备
精密称定适量绿原酸、咖啡酸、异槲皮苷、异绿原酸b、异绿原酸a、异绿原酸c等对照品,加甲醇分别定容于5ml棕色容量瓶,配制成绿原酸3.008mɡ/ml、咖啡酸2.096mɡ/ml、异槲皮苷2.03mɡ/ml、异绿原酸b3.04mɡ/ml、异绿原酸a3.04mɡ/ml、异绿原酸c3.048mɡ/ml的标准溶液。精密移取以上标准溶液配制成含绿原酸0.5998mɡ/ml、咖啡酸0.0503mɡ/ml、异槲皮苷0.0698mɡ/ml、异绿原酸b0.4001mɡ/ml、异绿原酸a0.8001mɡ/ml、异绿原酸c0.6005mɡ/ml的混合对照品溶液。
2.4色谱条件的建立
2.4.1流动相的选择
采用六种不同的流动相系统甲醇-水、乙腈-水、甲醇-0.05%磷酸水、甲醇-0.1%磷酸水、乙腈-0.1%磷酸水、乙腈-0.2%磷酸水等,当采用乙腈-0.1%磷酸水作为流动相系统,由于uplc仪器具有超高压的特点,致使色谱峰出现严重拖尾,为优化峰形,采用高酸度系统乙腈-0.2%磷酸水流动相系统,结果显示,乙腈-0.2%磷酸水洗脱出来的色谱峰峰形好,用时短,因此,后续试验采用乙腈-0.2%磷酸水作为流动相。
2.4.2洗脱程序的选择
本试验优选出三个不同的洗脱梯度进行比较,结果显示,其中梯度2洗脱出的hplc色谱峰在10~12min,色谱峰分离度较好、基线平滑,用时短,所以,选择梯度2作为后续试验用洗脱程序。
2.4.3柱温的选择
本试验考察30℃,35℃,40℃三个不同柱温,结果显示,当柱温为35℃时,色谱图中的各个峰出峰相当,分离度好,对色谱柱寿命无影响,因此,选择35℃为检测柱温。
2.4.4高中低流速的选择
本试验分别考察0.2ml/min、0.3ml/min、0.4ml/min低中高三个流速,结果显示,同一梯度下流速对色谱图的影响比较大,速度过大导致邻近色谱峰重合,0.2ml/min流速下的色谱图分离度适宜,用时短,因此,选择0.2ml/min作为测定流速。
2.4.5检测波长的选择
本试验对厚藤药材样品的hplc全谱进行全波长扫描,结果表明,254nm波长下色谱峰对多数物质有较大吸收,因此,后续试验采用254nm。
2.4.6进样量的选择
本试验比较了不同进样量,结果表明,进样1μl,色谱图的基线随时间向下倾斜,基线不平;进样3μl,色谱柱过载,色谱峰严重拖尾,进样2μl较为合适。
2.5指纹图谱的方法学考察
2.5.1精密度试验
精密称取厚藤(ip-5)药材粉末,照“2.2”项下方法制备供试品溶液,按照“2.1”项下色谱条件连续进样6次,检测指纹图谱,选取16个分离度>1.5的色谱峰计算相对峰面积,采用国家药典委员会组织开发的《中药色谱指纹图谱相似度评价系统》(v2.0)计算相似度,结果如表10~12所示,同一编号色谱峰在不同进样时间的相对峰面积的相对标准偏差rsd<3.90%,相对保留时间rsd≦0.30%,六次进样色谱图与共有模式的相似度范围是0.998~1.000,说明仪器的精密度合格,符合指纹图谱测定的要求。
表10精密度试验-相对峰面积(n=6)
表11精密度试验-相对保留时间(n=6)
表12精密度试验相似性结果
2.5.2重复性试验
精密称取同一批样品各6份,分别制备供试品溶液,依次进样,选取16个分离度>1.5的色谱峰计算相对峰面积,采用国家药典委员会组织开发的《中药色谱指纹图谱相似度评价系统》(v2.0)计算相似度,结果显示,各色谱峰相对峰面积的相对标准偏差rsd<3.20%,相对保留时间的相对标准偏差rsd<0.20%,各样品色谱图与共有模式的相似度范围是0.999~1.000,表明该方法的重复性好,符合hplc指纹图谱测定的要求。
2.5.3稳定性试验
取同一供试品溶液,分别在0h,2h,4h,8h,i2h,24h时进样,检测指纹图谱,选取16个分离度>1.5、峰面积较大的色谱峰计算相对峰面积,采用国家药典委员会组织开发的《中药色谱指纹图谱相似度评价系统》(v2.0)计算相似度,结果显示,各色谱峰相对峰面积的相对标准偏差rsd<4.16%,相对保留时间rsd<1.50%,不同时间点色谱图与共有模式的相似度范围是0.996~1.000,说明样品在该方法下,24h内成分稳定,符合hplc指纹图谱测定的要求。
2.6结合化学计量学建立厚藤的uplc指纹图谱
2.6.1色谱峰的指认
按“2.2”项下方法制备样品,取“2.3”项下混合对照品溶液,采用“2.1”项下色谱条件进样,根据相对保留时间tr,色谱峰光谱图一致性原则,从16个共有峰中指认六个峰,即编号峰4、5、9、11,12、14分别为绿原酸、咖啡酸、异槲皮苷、异绿原酸b、异绿原酸a、异绿原酸c,结果如图26所示。
2.6.2参照峰的确定
本试验选择分离度较好、峰面积较大、保留时间适宜的12号色谱峰(异绿原酸a)作为参照峰s。
2.6.3共有指纹峰的指定及相似度评价
10批厚藤药材共有峰编号图19,本试验采用国家药典委员会组织开发的《中药色谱指纹图谱相似度评价系统》(v2.0)评价10批厚藤药材的色谱图相似度,以ip-3为参照峰,采用中位数法生成对照图谱,计算相似度,10批厚藤药材与与共有模式的相似度值范围在0.945~1.000之间,结果如表13~14。
表1310批厚藤药材的相对峰面积
表1410批厚藤药材的相似度结果
2.6.4厚藤指纹图谱的聚类分析(hca)
聚类分析(hierarchicalclusteringanalysis)是常用的探索性统计方法,适用于分析受多因素影响的样本,目前在中药真伪鉴别、质量评价、品种分类等方面广泛应用。本试验将10批不同产地厚藤药材的16个共有峰的相对峰面积作为变量,得到10×16阶原始数据矩阵,导入spss19.0软件,采用离差平方和法(ward法),选用欧氏距离(euclidean)为测度,对数据进行标准化转换后进行q型聚类分析以挖掘厚藤指纹图谱数据,得到厚藤药材的聚类分析谱图,如图22,聚类分析将10批厚藤分为了3类,ⅰ类包括第3、4、5、6批,ⅱ类包括第2、9批,iii类包括第1、7、8、10批次。
2.6.5厚藤指纹图谱的主成分分析(pca)
主成分分析法(principalcomponentsanalysis)是将多个变量转化为少数的独立综合变量的降维分类分析法。主成分个数的选择原则包括:保留累计贡献率大于70%的主成分;保留特征值大于1的主成分。本试验将10批不同产地厚藤药材的16个共有峰的相对峰面积作为变量,对数据降维后进行因子分析。
表15主成分分析解释总变量
结果如表15所示,前四个主成分的特征值均大于1,第一主成分方差贡献率为41.885%,第二主成分方差贡献率为23.228%,第三主成分方差贡献率为12.911%,第四主成分方差贡献率为7.603%;pca1、pca2、pca3、pca4的累计贡献率为85.627%,可表达厚藤药材的大部分信息。
表16旋转成份矩阵
第一主成分的特征值最大,可较为全面地描述指纹图谱的信息,根据主成分分析的成分旋转成分矩阵表,其中色谱峰峰号4,11,14与第一主成分的相关性系数分别为0.873、0.886、0.944,表明该3个峰对厚藤药材的质量控制有重要意义,其中峰号4、11、14已被指认为绿原酸、异绿原酸b、异绿原酸c。
以pca1,pca2,pca3得分作图,10批厚藤药材分为3,4,5,6及1,2,7,8,9,10两类;以pca1,pca2,pca4得分作图,分为2,9及1,3,4,5,6,7,8,10两类,见图23~24,所得到的分类结果与聚类分析一致。
3.讨论与小结
本试验为了比较uplc指纹图谱与hplc指纹图谱,uplc指纹图谱中样品制备及对照品配制与hplc指纹图谱章节下的样品制备、对照品配制方法保持一致。
本试验采用相似度软件,生成10批次药材图谱的共有模式,生成ⅰ类、ⅱ类及ⅲ类厚藤液相色谱图的共有模式,第3、4、5、6批与10批次药材共有模式r的相似度均大于0.945,第2、9批次与10批次药材共有模式图谱r’的相似度均大于0.958,第1、7、8、10批与10批次药材共有模式图谱r’的相似度均大于0.962,根据进一步相似度分析结果,ⅰ类药材(即第3、4、5、6批)与其相应共有模式图谱r’1的相似度均大于0.981,ⅱ类药材(即第2、9批)与其相应共有模式r’2的相似度均大于0.979,ⅲ类药材(第1、7、8、10批)与10批次药材共有模式图谱r’3的相似度均大于0.967,根据以上结果可知,ⅰ类、ⅱ类及ⅲ类厚藤药材与共有模式r’1、r’2、r’3的相似度较10批次药材共有模式图谱r’更高,可更好地反映各批次厚藤的整体信息,为厚藤药材的质量控制提供依据。
本试验对10批厚藤药材的16个共有峰进行聚类分析及主成分分析,色谱峰4、11、14分别为绿原酸、异绿原酸b、异绿原酸c,是直接影响厚藤药材的分类、品质优劣的关键色谱峰。根据聚类分析结果:ⅰ类药材分别产自广西城港市江山半岛(脯鱼湾)、城港市江山半岛(白浪滩)、城港市江山半岛(白沙湾)、城港市北仑河口红树林自然保护区,ⅱ类药材分别产自防城港市江山半岛(三块石)、钦州市三娘湾,ⅲ类药材产自广西城港市防城区、广西北海市合浦县白沙镇独山村、海南陵水黎族自治县陵水英州镇清水湾、海南海口市秀英区,在色谱峰信息较多的情况下,药材分类与产地有关联,如ⅰ类药材均产自防城港市,其他类别药材受采收季节影响较大,无显著关联。
本试验用《中药色谱指纹图谱相似度评价系统》(v2.0)及spss19.0建立了uplc指纹图谱,结果显示,厚藤的uplc指纹图谱提供更多色谱峰信息,聚类分析及主成分分析的分类结果较hplc指纹图谱更详细,将广西及海南10批厚藤分为三类,采用相似性软件生成了3个共有模式,为厚藤药材提供了更为科学、合理、详实的质量控制参数。
- 还没有人留言评论。精彩留言会获得点赞!