一种板蓝根的质量控制方法与流程
本发明涉及药物化学成分检测技术领域,尤其涉及一种板蓝根的质量控制方法。
背景技术:
十字花科植物菘蓝Isatis indigoticaFort.的干燥根。秋季采挖,除去泥沙,晒干。分布于内蒙古、陕西、甘肃、河北、山东、江苏、浙江、安徽、贵州等地,常为栽培。具有清热解毒,凉血利咽的功效。用于温疫时毒,发热咽痛,温毒发斑,痄腮,烂喉丹痧,大头瘟疫,丹毒,痈肿。现代研究表明板蓝根具有抗菌、抗病毒、抗炎、节免疫等作用。其抗病毒、抗炎活性成分为氨基酸,抗菌、抗内毒素活性成分为有机酸类;板蓝根多糖在特异性、非特异性免疫、体液免疫和细胞免疫方面均有促进作用。《中国药典》(2010年版)对板蓝根药材中(R,S)-告依春的含量进行了控制。有文献报道采用HPLC法测定板蓝根中对(R,S)-告依春、靛玉红、腺苷、氨基酸等含量的研究。
已报道的板蓝根化学成分有:有机酸类、木脂素类、生物碱类、甾醇类、芥子苷类、含硫类化合物、多种氨基酸、核苷和多糖类。另外还含有生物碱类化合物包括吲哚类生物碱,如羟基靛玉红、依靛蓝酮等;喹唑酮类生物碱及其他类型生物碱。板蓝根也为疏风解毒胶囊中的重要组成药材,因此准确的确定该药材的药用物质基础、质量控制,对复方疏风解毒胶囊的药效贡献奠定基础和数据支撑。
技术实现要素:
本发明的目的是为了解决现有技术中板蓝根化合物检测不准确的缺点,而提出的一种板蓝根的质量控制方法。
为了实现上述目的,本发明采用了如下技术方案:
一种板蓝根的质量控制方法,包括以下步骤:
步骤一:对照品溶液制备:
对照品包括:精氨酸,胞苷,尿苷,腺苷,鸟苷,苯丙氨酸,告依春,异金雀花素;
准确称量2.00mg的上述对照品,分别置于10mL量瓶中,加入适量50%甲醇溶解,并以50%甲醇定容,得到质量浓度为200μg/mL对照品贮备液,进样前将对照品贮备液用50%甲醇稀释10倍;
步骤二:供试品溶液制备:
取板蓝根粉末1g,加入30%甲醇20mL置圆底烧瓶中,称定重量,超声提取45分钟,放冷,称定重量,并用70%甲醇补重,摇匀,经0.45μm微孔滤膜滤过,取续滤液备用;
步骤三:化合物测定:
通过HPLC-TOF/MS正负总离子流色谱图检测对照品溶液、供试品溶液,进行对比分析得出结果。
优选的,步骤三中:
色谱条件:色谱柱为Diamonsil II C 18色谱柱(250mm×4.6mm,5μm);流动相为乙腈-0.05%甲酸溶液,流动相梯度洗脱,体积流量为1.0mL/min;柱温35℃;进行200~600nm全扫;进样量5μL;
质谱条件:分流比设为1:4,TOF-MS实验条件:正负离子分别进行全扫描(ESI),扫描质量范围50~1200Da;干燥气的体积流量6L/min,干燥气温度180℃,雾化气压0.8Bar,阳离子模式下,毛细管电压4500V,阴离子模式下的毛细管电压2600V,碎裂电压200Vpp,选择甲酸钠溶液为内标矫正。
一种板蓝根的质量控制方法,包括以下步骤:
步骤一:对照品溶液制备:
对照品包括:精氨酸,胞苷,尿苷,腺苷,鸟苷,苯丙氨酸,告依春,异金雀花素;
准确称量2.00mg的上述对照品,分别置于10mL量瓶中,加入适量50%甲醇溶解,并以50%甲醇定容,得到质量浓度为200μg/mL对照品贮备液,进样前将对照品贮备液用50%甲醇稀释10倍;
步骤二:供试品溶液制备:
取板蓝根药材粉碎过40目筛,取粉末1g,精密称定,加入50%甲醇20mL,置具塞锥形瓶中,称定重量,超声提取45分钟,冷却至室温,再称定重量,用50%甲醇补足减失的重量,摇匀滤过,取续滤液,即得。
步骤三:化合物测定:
通过HPLC-TOF/MS正负总离子流色谱图检测对照品溶液、供试品溶液化合物种类,进行对比分析得出结果。
优选的,步骤三中:
色谱条件为:色谱柱:Orca C 18 5μ250*4.6mm;检测波长:220nm;柱温:35℃;流速:1mL/min;流动相:A-乙腈,B-0.05%磷酸;洗脱方式:梯度洗脱0~20min,0%~12%A,20~70min,12%~35%A;进样量:20μL;
质谱条件:分流比设为1:4,TOF-MS实验条件:正负离子分别进行全扫描(ESI),扫描质量范围50~1200Da;干燥气的体积流量6L/min,干燥气温度180℃,雾化气压0.8Bar,阳离子模式下,毛细管电压4500V,阴离子模式下的毛细管电压2600V,碎裂电压200Vpp,选择甲酸钠溶液为内标矫正。
本发明提出的一种板蓝根的质量控制方法,有益效果在于:本研究建立的板蓝根药材指纹图谱方法稳定、可靠、重现性好,可为败酱草药材的质量评价提供了依据。为疏风解毒胶囊质量的稳定均一性和疗效提供了保障。
附图说明
图1为本发明精密度试验的HPLC色谱图叠加图;
图2为本发明稳定性试验的HPLC色谱图;
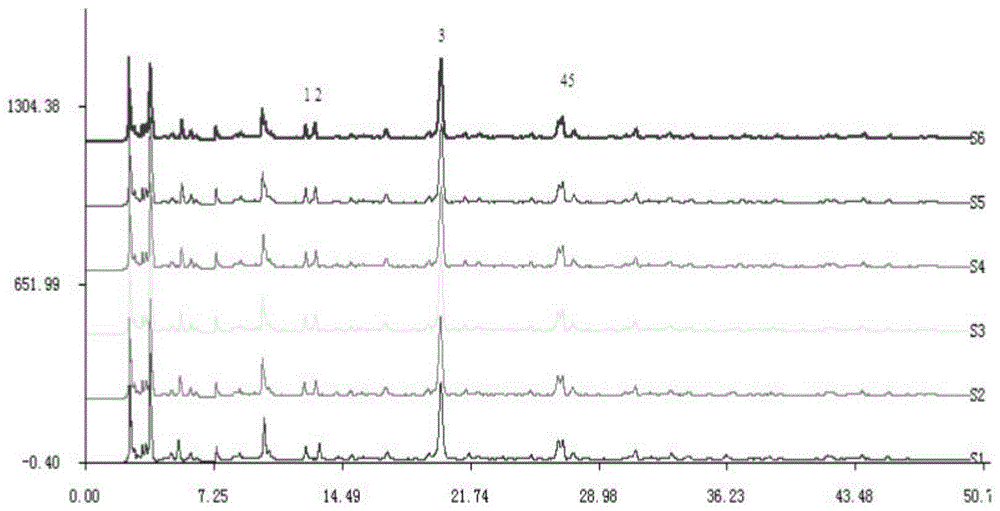
图3为本发明重复性试验的HPLC色谱图叠加图。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。
实施例一:
一种板蓝根的质量控制方法,包括以下步骤:
步骤一:对照品溶液制备:
对照品包括:精氨酸,胞苷,尿苷,腺苷,鸟苷,苯丙氨酸,告依春,异金雀花素;
准确称量2.00mg的上述对照品,分别置于10mL量瓶中,加入适量50%甲醇溶解,并以50%甲醇定容,得到质量浓度为200μg/mL对照品贮备液,进样前将对照品贮备液用50%甲醇稀释10倍;
步骤二:供试品溶液制备:
取板蓝根粉末1g,加入30%甲醇20mL置圆底烧瓶中,称定重量,超声提取45分钟,放冷,称定重量,并用70%甲醇补重,摇匀,经0.45μm微孔滤膜滤过,取续滤液备用;
步骤三:化合物测定:
通过HPLC-TOF/MS正负总离子流色谱图检测对照品溶液、供试品溶液,进行对比分析得出结果。
色谱条件:色谱柱为Diamonsil II C 18色谱柱(250mm×4.6mm,5μm);流动相为乙腈-0.05%甲酸溶液,流动相梯度洗脱,体积流量为1.0mL/min;柱温35℃;进行200~600nm全扫;进样量5μL;
质谱条件:分流比设为1:4,TOF-MS实验条件:正负离子分别进行全扫描(ESI),扫描质量范围50~1200Da;干燥气的体积流量6L/min,干燥气温度180℃,雾化气压0.8Bar,阳离子模式下,毛细管电压4500V,阴离子模式下的毛细管电压2600V,碎裂电压200Vpp,选择甲酸钠溶液为内标矫正。
通过HPLC-TOF/MS正负总离子流色谱图,共检测出23个离子流峰,识别了其中22个化合物。包括8个氨基酸类成分,1个生物碱类成分,3个糖类成分,3个木质素类成分,3个黄酮类成分和4个小分子酚酸类成分。其中3种化合物采用对照品进行确认。其中指纹图谱中主要特征峰为腺苷、次黄嘌呤、苯丙氨酸、鸟嘌呤衍生物等。各化合物的精确分子质量、多级质谱及PDA光谱信息见下表1
表1板蓝根液质指认化合物结构信息表
板蓝根液质指认化合物结构信息表
实施例二:
一种板蓝根的质量控制方法,包括以下步骤:
步骤一:对照品溶液制备:
对照品包括:精氨酸,胞苷,尿苷,腺苷,鸟苷,苯丙氨酸,告依春,异金雀花素;
准确称量2.00mg的上述对照品,分别置于10mL量瓶中,加入适量50%甲醇溶解,并以50%甲醇定容,得到质量浓度为200μg/mL对照品贮备液,进样前将对照品贮备液用50%甲醇稀释10倍;
步骤二:供试品溶液制备:
取板蓝根药材粉碎过40目筛,取粉末1g,精密称定,加入50%甲醇20mL,置具塞锥形瓶中,称定重量,超声提取45分钟,冷却至室温,再称定重量,用50%甲醇补足减失的重量,摇匀滤过,取续滤液,即得。
步骤三:化合物测定:
通过HPLC-TOF/MS正负总离子流色谱图检测对照品溶液、供试品溶液化合物种类,进行对比分析得出结果。
色谱条件为:色谱柱:Orca C 18 5μ250*4.6mm;检测波长:220nm;柱温:35℃;流速:1mL/min;流动相:A-乙腈,B-0.05%磷酸;洗脱方式:梯度洗脱0~20min,0%~12%A,20~70min,12%~35%A;进样量:20μL;
质谱条件:分流比设为1:4,TOF-MS实验条件:正负离子分别进行全扫描(ESI),扫描质量范围50~1200Da;干燥气的体积流量6L/min,干燥气温度180℃,雾化气压0.8Bar,阳离子模式下,毛细管电压4500V,阴离子模式下的毛细管电压2600V,碎裂电压200Vpp,选择甲酸钠溶液为内标矫正。
1、精密度试验
按样品制备方法制备板蓝根药材供试品溶液,在确定的色谱条件下测定,连续进样6次,测定HPLC色谱图,以3号峰的保留时间和色谱峰面积为参照,计算出各色谱峰的相对保留时间和相对峰面积。精密度试验的HPLC色谱图见图1,精密度试验数据见表2、3。可见,各色谱峰的相对保留时间及相对峰面积的RSD值均小于5%,符合指纹图谱的要求。
表2精密度试验相对保留时间数据
表3精密度试验相对峰面积数据
2、稳定性试验
取精密度下的供试品溶液,密闭,放置于室温,分别在0,3,6,9,12,24h时间间隔下检测指纹图谱,以3号峰的保留时间和色谱峰面积为参照,计算出各色谱峰的相对保留时间和相对峰面积。稳定性试验的HPLC色谱图见图2,由表4、5。可见,各色谱峰的相对保留时间及相对峰面积的RSD值均小于5%,符合指纹图谱的要求。说明供试品溶液在24h内稳定。
表4稳定性试验相对保留时间结果
表5稳定性试验相对峰面积结果
3、重复性试验
按确定的供试品溶液制备方法制备6份供试品溶液,在确定的色谱条件下测定,记录色谱图,以3号峰的保留时间和色谱峰面积为参照,计算出各色谱峰的相对保留时间和相对峰面积。重复性试验的HPLC色谱图见图3,重复性试验结果见表6、7,可见,各色谱峰的相对保留时间及相对峰面积的RSD值均小于5%,符合指纹图谱的要求。
表6重复性试验相对保留时间结果
表7重复性试验相对峰面积结果
综上所述,本研究建立的板蓝根药材指纹图谱方法稳定、可靠、重现性好,可为败酱草药材的质量评价提供了依据。为疏风解毒胶囊质量的稳定均一性和疗效提供了保障。
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,根据本发明的技术方案及其发明构思加以等同替换或改变而得到的技术方案、构思、设计,都应涵盖在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!