基于微藻蛋白质特征序列标签匹配的蛋白质快速检测方法及系统与流程
本发明属于微藻基因开发利用技术领域,特别是涉及一种基于微藻蛋白质特征序列标签匹配的蛋白质检测方法及系统。
背景技术:
海洋微藻是水体生态系统中主要的初级生产者,占海洋生物物种的40.86%,有体积小、数量大、细胞结构简单、生长周期短、生物量大、光合效率高、适应力强、不受气候限制、易培养等优点,因此受到了人们的广泛关注,微藻中所含有的微藻不饱和脂肪酸具有维护生物膜的结构和功能、离子通道的调节、调控基因的表达、参与活性物质的合成、预防和治疗心血管疾病、调节免疫系统功能等生理功能和生物学效应。脂肪酸去饱和酶是催化脂肪酸链特定位置形成双键,从而产生不饱和脂肪酸的一类酶类大家族。
由于对相关酶的蛋白质序列进行逐条分析时,可以针对其蛋白质序列中的某功能域进行分析,因此提取目标蛋白质的特征序列,并对其进行特征序列标签的匹配和搜索可提高获得目标蛋白质序列的效率。
蛋白质组学是了解基因功能的最重要的途径之一。而蛋白质组学研究中一项基本而又尤为重要的一步就是蛋白质序列的鉴定,也就是确定蛋白质的一级结构,即一个氨基酸串。现今存在有20种氨基酸,并且每个蛋白质的一级结构均能表示为一个由20种氨基酸组成的序列,每个氨基酸用固定字母表示。由于序列决定结构,结构决定功能,因此研究蛋白质序列是生物信息学领域的一个关键问题,为进一步研究蛋白质的功能提供依据,需要有效的序列分析方法。
蛋白质序列搜索是蛋白质科学的重要研究方向,研发速度快和精度高的鉴定算法是该方向的一个主要问题。确定蛋白质功能最可靠的方法是进行数据库的相似性搜索。现有方法的搜索结果只注明目标序列的匹配部分,并不能提供该序列的全部信息,有时甚至得出模棱两可的结果,对用户产生误导,且都需要辅以人工搜索,面对大量数据集时耗时长。
因此,为了准确、快速的在大量蛋白质组数据中搜索到所需要的目标序列,需要一种可进行大数据处理,快速提升检索速度,有效的在大量蛋白质序列组中对目标蛋白质序列进行特征序列标签的匹配和搜索的方法,达到提高搜索结果速度的目的以对脂肪酸去饱和酶序列进行分析。
技术实现要素:
本发明的目的是提供一种基于微藻蛋白质特征序列标签匹配的蛋白质快速检测方法及系统,以克服现有技术的不足。
基于微藻蛋白质特征序列标签匹配的蛋白质快速检测方法,其特征在于包括以下步骤:
步骤1:采集微藻样品,对样品进行RNA提取,样品纯度要求:OD值应在1.8至2.2 之间;电泳检测28S:18S至少大于1.8,样品浓度:总RNA浓度不低于400ng/μg,用①Oligo(dT)富集mRNA,去除rRNA,将RNA随机打断,用随机引物和逆转录酶从RNA片段合成cDNA片段,cDNA片段末端修复,连接测序接头;②对于small RNA,进行3’和5’端接头连接,逆转录合成cDNA片段,最终将①和②的cDNA片段制备测序文库并进行测序,获得转录组测序的基因序列,之后翻译为蛋白质序列集;
或者根据所采集微藻的种类同时从美国国家生物技术信息中心(NBCI)下载对应的蛋白质序列集,并与上述蛋白质序列集合并;
步骤2:对步骤1得到的蛋白质序列集进行预处理,得到标准蛋白质序列集;
所述蛋白质序列集中的蛋白质序列有几百万条,得到标准蛋白质序列之后,该标准蛋白质序列中的每一条可以由几个、几百个甚至更多氨基酸组成,每一个氨基酸以20个字母G、S、A、T、V、D、N、L、I、E、Q、Y、F、H、P、M、W、K、C、R中的其中一个来表示;
步骤3:对该标准蛋白质序列集进行拆分配置处理,将其转换为对于一个给定序列片段的键/值对列表;
(1)选择需要进行匹配检测的蛋白质Pr,该Pr作为目标蛋白质;
(2)基于已有的文献确定(1)中Pr的氨基酸序列特征值,该特征值由多个氨基酸组构成,每个氨基酸组中有多个氨基酸;以i表示组数,将特征值的每组氨基酸记为PCF1、PCF2...PCFi;
(3)根据特征值将步骤2的标准蛋白质特征序列集进行排列:
排列后的数据记作以下形式:
<PCF1,PLT1>;.......<>......<PCF1,PLTj>
.......
<PCFi,PLT1>;.......<>......<PCFi,PLTj>
其中,PCF1-PCFi表示(2)中的特征值,i表示组数,PLT1-PLTj表示步骤2的蛋白质序列集中的每一条蛋白质序列数据,j表示步骤2中标准蛋白质序列集所含蛋白质序列数据的数量;从而得到一个对列表;
(4)检测(3)中对列表的每一行PLT1-PLTj是否含有该行之前所标注的PCFk,k属于1-i,将该行包含有PCFk的那些PLT1-PLTj中的元素检测出来,记作
<PCF1,PL1-1>;.......<>......<PCF1,PL1-j>
.......
<PCFi,PL i-1>;.......<>......<PCFi,PLi-j>
并将记作上述格式的数据作为键/值对列表,
其中,PL1-1至PL1-j表示第一行PLT1-PLTj中含有PCF1的标准蛋白质序列,PL1-1表示PLT1-PLTj中第一个含有该PCF1的标准蛋白质序列,PL1-j表示PLT1-PLTj中最后一个含有该PCF1的标准蛋白质序列;PLi-1至PLi-j表示第i行PLT1-PLTj中含有PCFi的标准蛋白质序列,PLi-1表示PLT1-PLTj中第一个含有该PCFi的标准蛋白质序列,PLi-j表示PLT1-PLTj中最后一个含有该PCFi的标准蛋白质序列;
步骤4:对上述键/值对列表,进行汇总;
接受上一步骤生成的键/值对列表,合并含有相同特征值的蛋白质序列,得到了分别包含第1至第i个蛋白质特征值的蛋白质序列,记作
<PCF1,PL[1-1....1-j]>
...
<PCFi,PL[i-1....i-j]>
其中,<PCF1,PL[1-1....1-j]>=<PCF1,PL1-1,....PL 1-j>
...
<PCFi,PL[i-1....i-j]>=<PCF1,PLi-1,....PL i-j>
并将记作上述格式的数据作为减化后的键/值对列表;
步骤5:对上一步得到的减化后的键/值对列表进行交集运算,以得出步骤1的蛋白质序列集中是否含有步骤3中指定的目标蛋白质;
[PCF1,...,PCFi]=PL[1-1...1-j]∩...∩PL[i-1...i-j],
即以交集PL[1-1...1-j]∩...∩PL[i-1...i-j]表示上述PL[1-1...1-j]—PL[i-1...i-j]中,同时含有PCF1-PCFi的那些蛋白质序列;
当结果非空,则确定该数据集某条数据含有特征蛋白质序列。
该对列表可能过于庞大,超过某台物理机的处理能力,需要将其拆分,将其拆分成64M大小的文件,每个文件可以作为一个计算任务。均匀发送到参与检索的计算节点上(物理计算机),例如有三台物理机参与检索计算任务。而生成的对列表文件大小为800G,则拆分原则为:800GB/64MB为拆分成的文件数。每个计算节点(物理计算机)分配的文件数为800GB/64MB/3。
上述步骤3在其步骤(3)之后如下:
(4A)对所得的上述对列表进行分解:将对列表分解为若干个64M的文件,每个文件是上述对列表的一个子集,即子对列表;
(5A)设置多个检测节点(采用物理计算机),将得到的子对列表分配至每个检测节点进行进一步检测;
(6A)每个检测节点检测其所分配的各个64M文件进行检测,以检测每一个子对列表中的每一行是否是否含有该行之前所标注的PCFk,k属于1-i;
所有检测节点完成检测时,即完成对(3)中对列表的每一行PLT1-PLTj是否含有该行之前所标注的PCFk,k属于1-i,将该行包含有PCFk的那些PLT1-PLTj中的元素检测出来,记作
<PCF1,PL1-1>;.......<>......<PCF1,PL1-j>
.......
<PCFi,PL i-1>;.......<>......<PCFi,PLi-j>
并将记作上述格式的数据作为键/值对列表,
其中,PL1-1至PL1-j表示第一行PLT1-PLTj中含有PCF1的标准蛋白质序列,PL1-1表示PLT1-PLTj中第一个含有该PCF1的标准蛋白质序列,PL1-j表示PLT1-PLTj中最后一个含有该PCF1的标准蛋白质序列;PLi-1至PLi-j表示第i行PLT1-PLTj中含有PCFi的标准蛋白质序列,PLi-1表示PLT1-PLTj中第一个含有该PCFi的标准蛋白质序列,PLi-j表示PLT1-PLTj中最后一个含有该PCFi的标准蛋白质序列。
上述步骤3-5中,由于每个计算节点(物理计算机)的配置可能不同(如CPU、内存、网络开销等)而导致计算能力不同,因此常常出现某个计算节点计算完毕,而其他计算节点仍然存在大量计算任务(计算任务可以看做每个64MB的文件)的情况,因此采用了对计算任务进行动态加载分配方法;
当有n个检测节点,分别为1-n,假设分别分配了S1,...,Sn个任务,且1-n个检测节点有一个完成任务,即剩余任务为0,并且各个节点当前完成任务数分别为s1,....,sn;
各个节点的剩余任务数为S1-s1,...,Sn-sn,在非零的剩余任务中选取值最大者,动态调配一个任务给已完成任务的节点,更新完成任务节点的任务分配数加一,更剩余任务最大节点的任务分配数减一,每次当有节点出现分配任务全部完成时,重复上述分配方式,直到所有计算任务完成。
步骤3-5中,所述的检测是选取PCF1,且PCF1的长度为L;在PLTn,n∈1-i中依次检索PCF1,含有PCF1片段的PLTn,与PCF1组成一个对应值<KEYPCF1,VALUEPLTn>;
对PCF2-PCFi依次重复上述步骤,并在步骤4中将所有获得的对应值进行整理获得键/值对列表。
上述步骤4中,由于运算是在多个物理计算机上分布实现的,因此在各物理机计算任务全部完毕之后,需要将多个计算机上的运算结果集进行汇总。会同时进行了三个处理:混洗,排序和搜索任务汇总。
混洗阶段,根据每个检测节点检测结果中的PCF值(PCF1-PCFi其中之一),将结果传输到一个汇总处理机上(具有同一个PCF值的搜索任务可能分布在不同的检测节点上,这一步结束后,位于不同检测节点的检测结果都传输到了这一个汇总处理机上);此外,在传送到搜索任务汇总处理机之前,在本地首先进行一次PCF检索结果的汇总,这样可以减少不必要的网络资源消耗,提高系统执行和传输效率。这个步骤中的文件传输可以使用HTTP协议。
排序阶段,排序和混洗是同时进行的,这个阶段将来自不同搜索任务文件具有相同PCF值的键/值对按照PCF1到PCFi的顺序进行排序。
搜索任务汇总阶段,搜索任务汇总处理任务服务器对通过混洗和排序后的键/值对进行汇总处理。得到了分别包含第1至第i个蛋白质特征值的蛋白质序列,记作
<PCF1,PL[1-1....1-j]>
...
<PCFi,PL[i-1....i-j]>
其中,<PCF1,PL[1-1....1-j]>=<PCF1,PL1-1,....PL 1-j>
...
<PCFi,PL[i-1....i-j]>=<PCF1,PLi-1,....PL i-j>
并将记作上述格式的数据作为减化后的键/值对列表。
本发明采用分布式计算框架方法运用到微藻蛋白质特征序列标签匹配中的快速检测系统及其方法,分布式计算框架可运行在大规模集群上,通过基于分布式计算框架框架设计自身业务所需的搜索任务拆分配置和搜索任务汇总实现方式,可处理大量数据的分布式并行计算过程。适合搜索任务拆分配置以及搜索任务汇总来处理的数据集仅需要满足一个基本要求:待处理的数据集可以分解成许多小的数据集,而且每个小的数据集可以完全并行地处理。蛋白质特征序列标签匹配中的快速检测系统所要实现的目标是在大量氨基酸序列数据集中进行蛋白质序列标签的匹配,该过程可以通过搜索任务拆分配置以及搜索任务汇总方法进行分解,获得许多颗粒度较小的数据集,这些数据集格式统一后,可以进行并行的计算处理,满足分布式计算框架的计算要求。本发明通过提取蛋白质序列特征将该分布式计算框架这种大数据集挖掘方法引入到蛋白质序列检索中,用来判断蛋白质的同源性和相似性程度,相比传统逐条数据进行对比的方式,算法的鉴定准确率和效率有显著提高。同时随着实验数据的成倍增长,该算法的执行时间也等比例增长。整个算法的单机运行时间复杂度为O(n),相比传统方式不仅大大提升了检索效率,而且算法效率较高。即使当数据样本持续增长,单机性能遇到瓶颈时利用分布式计算框架分布式多机并行处理,实现了分布式数据处理,也大大提升整个系统的处理效率。该方法不仅具有良好的实用性,而且算法的时间复杂度低,性能高,对于蛋白质组学的研究具有重要意义。
附图说明
图1是本发明的检测系统的示意图。
图2是本发明的含任务分解/分配子模块的检测系统示意图。
图3是搜索任务拆分配置以及搜索任务汇总计算原理图。
图4是经过预处理后待检索的几十万至几百万蛋白质部分序列片段。
图5是单机模式下搜索任务拆分配置以及搜索任务汇总控制台输出情况示意图。
图6是检测最终结果。
图7是时间效率曲线。
其中,1、权限识别模块,2、检测数据输入模块,21、目标蛋白质输入子模块,22、特征值输入子模块,23、蛋白质序列集输入子模块,24、NBCI蛋白质序列集输入子模块,3、蛋白质序列集汇总模块,4、键/值对列表模块,5、数据检测模块,6、特征值交集运算模块,7、检测输出模块,8、任务分解/分配子模块,9、检测节点,10、汇总处理机,11、汇总子模块。
具体实施方式
如图1所示,与上述方法相对应,本发明的基于微藻蛋白质特征序列标签匹配的蛋白质快速检测系统,其特征在于该系统包括:
权限识别模块1,该权限识别模块1通过验证所输入的操作员代码及其密码以判断该操作员是否有权限使用本系统;
与上述权限识别模块1相连的检测数据输入模块2,包括目标蛋白质输入子模块21、特征值输入子模块22、蛋白质序列集输入模块23、NBCI蛋白质序列集输入子模块24;
其中,所述目标蛋白质输入子模块21与特征值输入子模块22相连,在向目标蛋白质输入子模块21输入目标蛋白质的名称之后,向所述的特征值输入子模块22输入目标蛋白质的特征值参数,参数包括特征值所含氨基酸组的个数i以及每个氨基酸组所包含的氨基酸片段;所输入的氨基酸片段表示为多个连续的字母,且每个字母为20个字母G、S、A、T、V、D、N、L、I、E、Q、Y、F、H、P、M、W、K、C、R中的其中一个;
所述的蛋白质序列集输入模块22,该模块应于输入翻译为蛋白质序列集的微藻基因序列;所述的NBCI蛋白质序列集输入子模块24用于输入从美国国家生物技术信息中心数据库对外开放接口获得的待检测蛋白质序列数据;
所述的检测数据输入模块2与蛋白质序列集汇总模块3相连,所述的蛋白质序列集汇总模块3将所述蛋白质序列集输入模块23与NBCI蛋白质序列集输入子模块24进行汇总,将蛋白质序列集的数量记为j,并将目标蛋白质特征值的i个氨基酸组进行排列,将排列后的氨基酸组标记为PCF1、PCF2...PCFi,将蛋白质序列集进行排列,并将排列后的蛋白质序列集标记为PLT1、PLT2...PLTj;(显然可以移去NBCI蛋白质序列集输入子模块24与蛋白质序列集汇总模块3,而是将蛋白质序列集输入模块23直接作为汇总数据来使用);
所述的蛋白质序列集汇总模块3与键/值对列表模块4相连,该键/值对列表模块4将经过蛋白质序列集汇总模块3排列后的氨基酸组和蛋白质序列集建立成以下形式的队列表:
<PCF1,PLT1>;.......<>......<PCF1,PLTj>
.......
<PCFi,PLT1>;.......<>......<PCFi,PLTj>
其中,PCF1-PCFi表示目标蛋白质的特征值,i组数;PLT1-PLTj表示蛋白质序列集中的每一条蛋白质序列数据,j表示标准蛋白质序列集所含蛋白质序列数据的数量;从而得到一个键/值对列表;
所述的键/值对列表模块4与检测识别模块5相连,该检测识别模块5接受上述键/值对列表模块4生成的键/值队列表,检测所得队列表每一行的PLT1-PLTj是否含有该行之前所标注的PCFk,k属于1-i,将该行包含有PCFk的那些PLT1-PLTj中的元素检测出来,记作
<PCF1,PL1-1>;.......<>......<PCF1,PL1-j>
.......
<PCFi,PL i-1>;.......<>......<PCFi,PLi-j>
并将记作上述格式的数据作为简化后的键/值对列表,
其中,PL1-1至PL1-j表示第一行PLT1-PLTj中含有PCF1的标准蛋白质序列,PL1-1表示PLT1-PLTj中第一个含有该PCF1的标准蛋白质序列,PL1-j表示PLT1-PLTj中最后一个含有该PCF1的标准蛋白质序列;
...
PLi-1至PLi-j表示第i行PLT1-PLTj中含有PCFi的标准蛋白质序列,PLi-1表示PLT1-PLTj中第一个含有该PCFi的标准蛋白质序列,PLi-j表示PLT1-PLTj中最后一个含有该PCFi的标准蛋白质序列;
所述的检测识别模块5与特征值交集运算模块6相连,
所述的特征值交集运算模块6对检测识别模块5得到的减化后的键/值对列表进行交集运算,以得出蛋白质序列集汇总模块3中的蛋白质序列集中是否含有特征值输入子模块22中蛋白质特征值;
[PCF1,...,PCFi]=PL[1-1...1-j]∩...∩PL[i-1...i-j],
即以交集PL[1-1...1-j]∩...∩PL[i-1...i-j]表示上述PL[1-1...1-j]—PL[i-1...i-j]中,同时含有PCF1-PCFi的那些蛋白质序列;
所述的特征值交集运算模块6与检测输出模块7相连,
当上述交集非空,则确定蛋白质序列集汇总模块3中的蛋白质序列集中含有目标蛋白质输入子模块21输入目标蛋白质,所述的检测输出模块7将该结果输出;
当上述交集为空集,则确定蛋白质序列集汇总模块3中的蛋白质序列集中不含有目标蛋白质输入子模块21输入目标蛋白质,所述的检测输出模块7将该结果输出。
如图2、3所述,在上述系统基础上进行改进,所述的检测识别模块5包括任务分解/分配子模块8、多个检测节点9、多个汇总处理机10和汇总子模块11,其中所述的任务分解/分配模块5相连,该任务分解/分配模块8将对列表数据拆分成多个64M大小的文件,每个文件均为对列表的一个子集,并将文件均匀发送到各个检测节点9上;
所述的检测节点9接受上述任务分解/分配子模块8分配的文件,检测所分配文件的每一行是否含有该行之前所标注的PCFk,k属于1-i;
所有检测节点完成检测时,即完成检测对列表的每一行PLT1-PLTj是否含有该行之前所标注的PCFk,k属于1-i,
每一个检测节点9都对应有一个汇总处理机10,首先由其中一个汇总处理机10将与之对应的检测节点9上的数据进行初步汇总;
当所有的汇总处理机10都完成汇总之后,由汇总子模块11对各个汇总处理机10中的数据进行进一步汇总;从而实现将对列表每一行中包含有该行之前所标注的PCFk的那些PLT1-PLTj中的元素检测出来,记作
<PCF1,PL1-1>;.......<>......<PCF1,PL1-j>
.......
<PCFi,PL i-1>;.......<>......<PCFi,PLi-j>
并将记作上述格式的数据作为键/值对列表,
其中,PL1-1至PL1-j表示第一行PLT1-PLTj中含有PCF1的标准蛋白质序列,PL1-1表示PLT1-PLTj中第一个含有该PCF1的标准蛋白质序列,PL1-j表示PLT1-PLTj中最后一个含有该PCF1的标准蛋白质序列;PLi-1至PLi-j表示第i行PLT1-PLTj中含有PCFi的标准蛋白质序列,PLi-1表示PLT1-PLTj中第一个含有该PCFi的标准蛋白质序列,PLi-j表示PLT1-PLTj中最后一个含有该PCFi的标准蛋白质序列。
如果检测所有的结果均为空集,表明该待检索的样本集中不包含特征值序列。
实施例
对于特征酶参数的输入,以如下的数据为例:
脂肪酸去饱和酶家族主要由delta-4、delta-5、delta-6、delta-8、delta-9和delta-12六种去饱和酶组成,每个脂肪酸去饱和酶序列包含多个特定的蛋白质特征片段。例如需要检索的为如下去饱和酶:
Delta-4:HPGG、HMGGH、HNKHH、QIEHH
Delta-5:HPGG、HEGGH、HNKHH、QIEHH
Delta-6:HDTLH、HNLHH、QIEHH
Delta-8:HPGG、HDYLH、HNTHH、QTEHH
Delta-9:HRTHH、HNWHH
Delta-12:HECGH、HAKHH、HVVHH
i∈1-15,蛋白质特征值片段(Protein Character Fragment)j定义为:
PCFi∈[HRTHH,HNWHH,HPGG,HEGGH,HNKHH,QIEHH,DHTLH,HNLHH,HECGH,HAKHH,HVVHH,HDYLH,HNTHH,QTEHH,HMGGH]
对于步骤1,获得的待检索文本文件往往几百兆甚至几个G,在大数据集中找出符合上述特征的delta序列,传统逐条检索的方式显然不能满足快速、高效匹配的要求。
对于步骤2,sequence文件由20个字母组成的蛋白质序列,对每种脂肪酸去饱和酶序列的检索即对文件中的特定字符串的检索。每个序列PLT均以符号“>”开始,至下一以“>”起始的序列结束,序列字符串由多行组成。
由于分布式计算框架进行数据处理默认以行为单位进行数据的输入,因此,需要将文件中由多行组成的序列整理为单行序列,并消除序列与序列之间的空白行。最终形成标准的以行为单位代表蛋白质序列的标准格式文件。
该案例选取了格式标准化转换后,数据集规模从60M到1G的数据进行测试。同时,由于分布式计算框架的运行效率与机器性能、多机之间通信等因素相关,为了减少客观因素对实验结果造成影响,该案例采取单机模式运行。
该数据集包含n行,设j∈1-n,蛋白质序列标签(Protein List Table)j定义为PLTj。
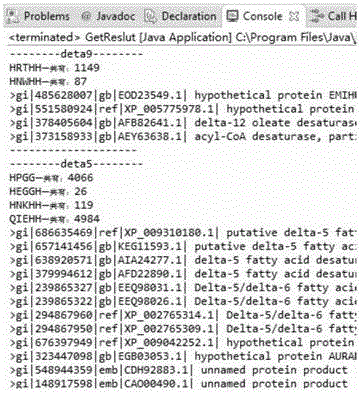
经过预处理后待检索的几十万至几百万蛋白质部分序列片段如图4所示:
对于步骤3,对该标准格式文件进行搜索任务拆分配置处理,接受标准格式的序列文件并将其转换为一个特定序列片段的键/值对列表。Mapper处理后输出文件的每个特征片段(键)对应文档中的相应序列(值):
<PCF1,PLT1>;.......<>......<PCF15,PLTj>
在搜索任务拆分配置操作后通过一个中间结果的数据集进行存储和处理搜索任务拆分配置部分和搜索任务汇总部分之间的通信。当搜索任务拆分配置模块的输出被收集后,它们会被以指定的方式区分地写出到输出文件里。我们可以为搜索任务拆分配置模块提供合并操作,在搜索任务拆分配置模块输出它的<key,value>时,键值对不会被马上写到输出里,他们会被收集在列表里(一个key值对应一个列表),当写入一定数量的键值对时,这部分缓冲会被合并操作中进行合并,然后再输出到相应的分区中。
对于步骤4,对该列表值集合,进行搜索任务汇总处理。接受上一步骤生成的列表,合并相同键值的蛋白质特定序列片段缩小键/值对列表。搜索任务汇总处理后得到了分别包含了15个蛋白质特征值片段的蛋白质序列标签数组。
<PCF1,PLT[1....j]>
...
<PCF15,PLT[1....j]>
这个阶段如果涉及多台机器参与,后台会同时进行了三个处理:混洗(Shuffle),排序(sort)和reduce。
混洗阶段,引入了搜索任务拆分配置以及搜索任务汇总框架会根据搜索任务拆分配置结果中的key值,将相关的结果传输到某一个搜索任务汇总处理上(多个搜索任务拆分配置项产生的同一个key的中间结果分布在不同的机器上,这一步结束后,他们传输都到了处理这个key的搜索任务汇总处理任务的机器上)。这个步骤中的文件传输可以使用HTTP协议。
排序和混洗是一块进行的,这个阶段将来自不同搜索任务拆分配置具有相同key值的<key,value>对合并到一起。
搜索任务汇总阶段,上面通过混洗和排序后得到的<key,(list="″of="″values)="″>会送到搜索任务汇总模块中处理,输出的结果通过格式化的输出文件,输出到分布式文件系统中。
单机模式下搜索任务拆分配置以及搜索任务汇总控制台输出如图5所示:
对于步骤5,6个脂肪酸去饱和酶:Delta-n(n∈[4、5、6、8、9、12])分别由包含多个特定蛋白质特征片段的序列组成,因此,通过对多个蛋白质序列标签数组进行交集运算就可以得出实验数据集中蛋白质序列属于某个特定的脂肪酸去饱和酶delta-n。
Delta-n=[PCF1...PCFi]=PLT[1..j]∩...∩PLT[1..j]
检测最终结果如图6所示:
两个蛋白质特征值片段HRTHH与HNWHH各1149和87个,通过AND运算后获得整个实验结果数据中满足delta-9特征的四个蛋白质序列标签。同样可以快速、准确地获得所有脂肪酸去饱和酶序列。
为了对比分析该蛋白质特征序列标签匹配的蛋白质的快速检测系统的检索效率,本实验分别选取了60M、90M、150M、300M、600M以及1G的Fasta格式数据进行检索通过比较程序执行时间从而得出本系统的检索性能。
检索效率分析表
算法时间效率曲线如图7所示:
通过算法时间效率曲线图可以看出,整个曲线几乎以固定夹角成一条直线,随着实验数据的成倍增长,该算法的执行时间也等比例增长。整个算法的单机运行时间复杂度为O(n),相比传统人工方式不仅大大提升了检索效率,而且算法效率较高。即使当数据样本持续增长,单机性能遇到瓶颈时利用分布式计算框架分布式多机并行处理也会大大提升整个系统的处理效率。
- 还没有人留言评论。精彩留言会获得点赞!