电子器件和化合物的制作方法
本发明涉及电子器件、电掺杂的半导体材料和用于所述电子器件的化合物。
背景技术:
在有机电子器件之中,有机太阳能电池(OSC),亦称有机光伏(OPV)器件,具有最多变的器件架构。通常,它们包含至少一个安排在两个电极之间的有机半导体层。所述有机层可以是供体和受体例如P3HT(聚3-己基噻吩)和PCBM(苯基C61丁酸甲酯)的掺合物。如果使用界面注入层促进电荷载流子注入/提取的话,这么简单的器件结构只适度地达到效率(Liao等,Appl.Phys.Lett.,2008.92:173303页)。其他有机太阳能电池具有多层结构,有时甚至混杂聚合物/小分子结构。串联或多单元堆叠体也是已知的(参见US 2007/090371 A1,或Ameri等,Energy&Env.Science,2009.2:347页)。因为不同的层可以包含适合于不同功能的不同化合物和它们的混合物,所以多层器件能更容易优化。典型的功能层是传输层、光活性层、注入层等。
在OPV中,光学活性化合物应理解是对太阳光谱的至少某个波长范围具有高吸光系数的化合物,所述化合物将吸收的光子转化为激子。激子可以解离为自由电荷,后者可以最终作为光电流提取。所述光活性化合物通常用于供体-受体异质结中,其中所述供体或受体的至少一种是光吸收化合物。所述供体-受体异质结的界面负责将产生的激子分离成电荷载流子。所述异质结可以是本体异质结(掺合物)、或平面(flat)(也称为平面(planar))异质结,也可以提供附加层(Hong等,J.Appl.Phys.,2009.106:064511页)。
高效OPV器件应最小化复合引起的激子损失。因此,所述异质结中的化合物必须具有高电荷载流子迁移率和高激子扩散长度。所述激子必须在异质界面处分离成电荷载流子并且所述电荷载流子必须在任何复合发生之前离开所述光学活性区。因此,目前,富勒烯(C60、C70、PCBM等等)是作为OPV器件中受体材料的优选选择。
光电子器件的电荷传输材料要求是透明的,至少在所述器件活性的波长内,并具有良好的半导体性质。这些半导体性质是内在的,例如能级或迁移率,或外来的,例如电荷载流子密度。所述电荷载流子密度也可以通过用电氧化还原掺杂剂掺杂所述化合物来受外在影响。
OSC常常需要在n-掺杂的电子传输层中使用至少一种n-掺杂剂,或作为纯夹层,促进电子从导电层注入半导体或从半导体注入另一种半导体。
各种强氧化还原n-掺杂剂是已知的,例如EP1768200B1中的四(1,3,4,6,7,8-六氢-2H-嘧啶并[1,2-a]嘧啶根合)二钨(II)、和双(2,2'-三吡啶)钌及其他。强还原型n-掺杂剂的一个主要问题是因为它们是强电子供体,所以它们容易通过与大气氧反应而降解。没有很多已知的化合物能够直接充当n-掺杂剂并又是空气稳定的。开发前体化合物目的在于提供空气稳定的有机化合物并且能够充当n-掺杂剂,这样的前体的例子公开在WO 2007/107306 A1中。
对于OSC中使用的低LUMO化合物,例如富勒烯(例如C60)或富勒烯衍生物(例如PCBM),包含离域电子的共轭体系的膦亚胺化合物在WO2012/175219中被证明是足够强的n-掺杂剂,并仍然是空气稳定的。然而,更广泛地提供用于有机电子器件的空气稳定并且足够强的n-掺杂剂、允许选择对于各种器件设计适当的掺杂剂以及制造程序的简化和高度再现性,特别是在大量生产中,仍然呈现出未满足的需求。
技术实现要素:
本发明的目的是提供具有良好的性能以及顺畅和可再现的加工性能的代用器件。本发明的另一个目的是提供代用的空气稳定的半导体材料和用于这样的材料的化合物。
所述目的由包含式1化合物的电子器件实现
ABx(1),
其中
A是由至少两个原子组成并包含离域电子的共轭体系的结构部分,
各B独立地选自亚胺官能团(Ia)
其中R1、R2、R3、R4独立地选自C1-C30烷基、C2-C30烯基、C2-C30炔基、C3-C30环烷基、C6-C30芳基、C2-C30杂芳基、C7-C30芳基烷基、C3-C30杂芳基烷基,
波形线表示与所述亚胺氮原子的共价键,
G在各基团(Ia)中独立地选自季碳原子和亚环丙烯基结构部分,
x是等于1或更高、优选等于2或更高的整数,和
所述亚胺氮原子的孤电子对和/或至少一个基团B的亚胺双键的π电子与所述结构部分A中包含的离域电子的共轭体系共轭,
条件是取代基R1、R2、R3、R4的两个或更多个可以相连以形成也可以含有不饱和度的环,并且如果取代基R1、R2、R3、R4的任何一个包含两个或更多个碳原子,则所述取代基中或由两个相连的取代基形成的任何环中的总体碳原子计数的最多三分之一能够被独立地选自O、S、N和B的杂原子置换。
优选地,x等于2或更高;更优选,x是选自2、3和4的整数。
还优选,所述亚胺氮原子的孤电子对和/或至少两个基团B的亚胺双键的π电子与所述结构部分A中包含的离域电子的共轭体系共轭。更优选地,所述亚胺氮原子的孤电子对和/或所有基团B的亚胺双键的π电子与所述结构部分A中包含的离域电子的共轭体系共轭。
更加优选地,A是含有一个离域电子共轭体系的C3-C40芳烃或C2-C40杂芳烃结构部分。
最优选地,A是C6-C18芳烃或C4-C18杂芳烃结构部分并且所述亚胺氮原子的孤电子对和/或所有基团B的亚胺双键的π电子与所述结构部分A中包含的离域电子的共轭体系共轭。
在优选实施方式中,所述电子器件是有机电子器件。
根据本发明的优选实施方式,式1的化合物用作n-掺杂剂。
根据优选实施方式,所述电子器件具有包含若干层的层状结构,其中至少一个所述层包含所述式1的化合物。所述电子器件还可以包含电子传输层。替代地或补充地,所述电子器件可以包含第一电极和/或第二电极。
在优选实施方式中,所述电子器件的包含式1化合物的层是电子传输层。更优选地,所述电子器件包含电子传输层,所述电子传输层包含形成均匀混合物的电子传输化合物和所述式1化合物。根据本发明的另一种优选方式,所述电子器件的包含式1化合物的层与电子传输层直接接触。在本发明的优选方式中,所述电子传输层包含富勒烯或富勒烯衍生物作为它的主要组分。
如果所述式1化合物纯态用作电子注入和/或提取层,所述电子器件的包含所述式1化合物的层优选具有小于5nm的厚度。
优选地,所述电子器件的包含所述式1化合物的层与电极、更优选阴极直接接触。附加或替代地,所述包含式1化合物的层安排在电子传输层和阴极之间。
在本发明的一个方面,所述电子器件包含连接单元(其还另称为pn-结或电荷发生层)。连接单元在串联器件中、例如串联OLED中或串联太阳电池中为形成所述串联器件的具体器件的电连接服务。在优选实施方式中,所述电子器件的包含式1化合物的层是所述连接单元的一部分。
在本发明的优选方式中,所述电子器件是太阳能电池,优选有机太阳能电池(OSC)。所述太阳能电池可以包含,例如,阳极、阴极和光吸收层。在优选实施方式中,所述有机太阳能电池还包含所述式1的化合物,其中所述化合物包含在所述光吸收层和阴极之间。在本发明的优选方面,所述有机太阳能电池包含pi、ni、或pin结构,其包含各第一p、i或n层。在此,p表示p-掺杂的空穴传输层,n表示n-掺杂的电子传输层,和i是内在光活性层(进一步的细节参见US 2007/090371 A1)。所述传输层具有比所述光活性层更大的HOMO-LUMO能隙(HOMO–最高占有分子轨道,LUMO–最低未占分子轨道)。
所述太阳能电池可以优先包含含有所述光吸收层的光吸收单元和包含附加光吸收层的附加光吸收单元。所述连接单元可以是连接所述光吸收单元与所述附加光吸收单元的pn-结。优选地,所述连接单元是在串联器件中或在多重堆叠的器件中连接所述光吸收单元与所述附加光吸收单元的pn-结。多重堆叠的器件是有三个或更多个光吸收单元的器件,有时也称为多级串联。多重堆叠的pin、pi或ni器件是优选的。附加或替代地,所述连接单元可以是连接所述阴极或阳极与所述光吸收单元的pn-结。
另一个目的通过包含至少一种电子传输基质化合物和至少一种具有式1的n-掺杂剂的电掺杂半导体材料实现
ABx(1),
其中
A是由至少两个原子组成并包含离域电子的共轭体系的结构部分,
各B独立地选自亚胺官能团(Ia)
其中R1、R2、R3、R4独立地选自C1-C30烷基、C2-C30烯基、C2-C30炔基、C3-C30环烷基、C6-C30芳基、C2-C30杂芳基、C7-C30芳基烷基、C3-C30杂芳基烷基,
条件是取代基R1、R2、R3、R4的两个或更多个可以相连以形成也可以含有不饱和度的环,并且如果取代基R1、R2、R3、R4的任何一个包含两个或更多个碳原子,则所述取代基中或由两个相连的取代基形成的任何环中的总体碳原子计数的最多三分之一能够被选自O、S、N和B的杂原子置换,
波形线表示与所述亚胺氮原子的共价键,
G在各基团(Ia)中独立地选自季碳原子和亚环丙烯基结构部分,和
x是等于1或更高的整数,
并且所述亚胺氮原子的孤电子对和/或至少一个基团B的亚胺双键的π电子与所述结构部分A中包含的离域电子的共轭体系共轭。
由等摩尔量的所述基质化合物和它的阴离子团组成的氧化还原对通过在与比较氧化还原对相同的条件下的循环伏安法测量的氧化还原电位值可以等于或高于由等摩尔量的四氰基醌二甲烷和它的阴离子团组成的所述比较氧化还原耦,然而,优选所述基质材料中包含的各化合物的氧化还原电位比比较氧化还原耦TCNQ/TCNQ阴离子团的氧化还原电位更负。
另外,术语“更负”将简化为“更低”。更优选地,所述基质材料的氧化还原电位低于-0.50V vs.标准氧化还原耦二茂铁鎓/二茂铁(Fc+/Fc),更加优选低于-0.65V vs.Fc+/Fc,更加优选低于-0.80V vs.Fc+/Fc,最优选=低于-0.90V vs.Fc+/Fc。
本发明的第三个目的由具有式1的化合物实现
ABx(1),
其中
A是由至少两个原子组成并包含一个离域电子共轭体系的结构部分,
各B独立地选自亚胺官能团(Ia)
其中R1、R2、R3、R4独立地选自C1-C30烷基、C2-C30烯基、C2-C30炔基、C3-C30环烷基、C6-C30芳基、C2-C30杂芳基、C7-C30芳基烷基、C3-C30杂芳基烷基,
波形线表示与所述亚胺氮原子的共价键,
G在各基团(Ia)中独立地选自季碳原子和亚环丙烯基结构部分,
x是等于2或更高的整数,和
所述亚胺氮原子的孤电子对和/或各基团B的亚胺双键的π电子与所述结构部分A中包含的所述离域电子的共轭体系共轭,
条件是如果G是季碳原子,或如果优选G是季碳原子或亚环丙烯基结构部分,则
R1、R2中的至少一个和R3、R4中的至少一个独立地选自C6-C30芳基和C2-C30杂芳基,或
R1与R2相连和R3与R4相连以形成也可以含有不饱和度的环,并且
如果取代基R1、R2、R3、R4的任何一个包含两个或更多个碳原子,则所述取代基中或由两个相连的取代基形成的任何环中的总体碳原子计数的最多三分之一能够被选自O、S、N和B的杂原子置换。
优选地,所述结构部分A选自C3-C40芳烃和C2-C40杂芳烃,更优选选自C6-C18芳烃和C4-C18杂芳烃。
发明的详细说明
本发明具有可通过掺杂用于OSC的典型电子传输基质(ETM)来实现高导电性的优点。利用所述式1的化合物,有可能取决于基质得到在10-2-10-6S/cm的期望范围内的电导率,只要掺杂剂浓度约10mol.%,如有机半导体中所最常用的。此外,所述式1的化合物具有高稳定性,允许它,例如,在真空中加工,例如通过真空热蒸发(VTE),或通过有机气相沉积(OVPD)。或者,所述式1的化合物可在惰性气氛下或甚至曝露在空气中通过溶液加工沉积。
在优选实施方式中,所述式1的化合物插入基质材料中形成掺杂层。由此,形成源自于所述式1化合物的分子的阳离子团,特别是通过从所述式1化合物传递至少一个电子到周围基质材料。在所述电子传递过程中,也形成所述基质材料的阴离子团。以这种方式,所述基质材料获得与未掺杂的基质材料的电导率相比增加的电子电导率。
所述基质材料可以由一种或多种基质化合物组成。所述电未掺杂的基质材料的电导率一般大约10-8S/cm或更低,特别是经常10-10S/cm左右。所述基质材料应该具有足够高的纯度。这样的纯度可利用常规方法、例如梯度升华达到。通过掺杂,所述掺杂的半导体材料的电导率可增加到超过10-6S/cm。OLED中使用的基质化合物它们的氧化还原电位(根据IUPAC惯例表达为由电中性基质分子和它的阴离子团组成的氧化还原对的电位)优选在-2.0和-3.0V之间的范围vs.Fc/Fc+。在OSC中,可用做电子传输基质的化合物具有小于-0.3V vs.Fc/Fc+的氧化还原电位,优选小于-0.8V vs.Fc/Fc+。符号Fc/Fc+涉及氧化还原对二茂铁/二茂铁鎓,其在整个本申请中用作等于零的参比氧化还原电位,因为所述Fc/Fc+耦在电化学电位测定、例如通过循环伏安法(CV)中最常用作标准参比氧化还原对。测定还原电位的循环伏安法和其他方法的细节以及二茂铁/二茂铁鎓参比耦与各种参比电极的关系可见于A.J.Bard等,“电化学方法:基本原理和应用(Electrochemical Methods:Fundamentals and Applications)”,Wiley,第2版,2000。
在本申请中,掺杂剂应被理解为混合在基质材料中的化合物(“基质材料被掺杂剂掺杂”)。在现有技术中也普遍使用术语“电掺杂剂”,或对于ETM的掺杂剂只是“n-掺杂剂”。
所述电子器件的安排在邻近于电子传输层的所述包含式1化合物的层可作为电子提取层用于OSC中。发现所述式1的化合物可在电子元件中,优选在电极和可以掺杂的半导体层之间用作电子注入层。替代或补充地,所述式1化合物可用作阻挡层,优选在吸收层和传输层之间,或用作电子元件中的半导体层。
在本发明的一个优选方面,所述电子器件的所有有机层都由小分子构成。优选地,所述小分子可通过VTE(真空热蒸发)沉积。
在本发明的另一个方面,至少一个有机半导体层包含聚合物,其中所述聚合物层和/或至少一个附加半导体层包含式1的化合物。
所述式1的化合物具有形成电导率比较高的很稳定的n-掺杂层的特殊优点。
如果所述式1的化合物在电子传输层中以与至少一种电子传输基质化合物的混合物用于太阳能电池中的话,所述电子传输层具有超过5nm、优选超过10nm、更优选超过30nm、更加优选超过50nm的厚度是有利的,特别是如果与经常具有高粗糙度的ITO阳极一起使用的话。如果所述包含式1化合物的ETL邻近于粗糙阴极、例如ITO阴极使用并且在所述粗糙阴极和所述ETL之间没有平滑夹层的话,使用厚达100nm、150nm或甚至更厚的ETL可能是有利的。
因为用于OSC中的很多电子传输基质化合物,例如富勒烯和它们的衍生物,具有显著的光学吸光度,其有利于它们在吸收层中的使用但不利于它们在传输层中的使用,因此本发明的化合物具有与这样的基质化合物相比较低的吸光度并且它们能以高浓度用于所述n-掺杂的电子传输半导体材料中是非常有利的,这减轻了在ETL中的不希望的、寄生光吸收。对于在厚ETL中、特别在厚于50nm的ETL中并且尤其在厚于100nm的ETL中应用,所述式1化合物以高于10wt%、更优选高于20wt%、并且甚至更优选高达30wt%或更高的浓度使用是有利的。
附图说明
下面,参考绘图公开示例性的实施方式。所述图显示:
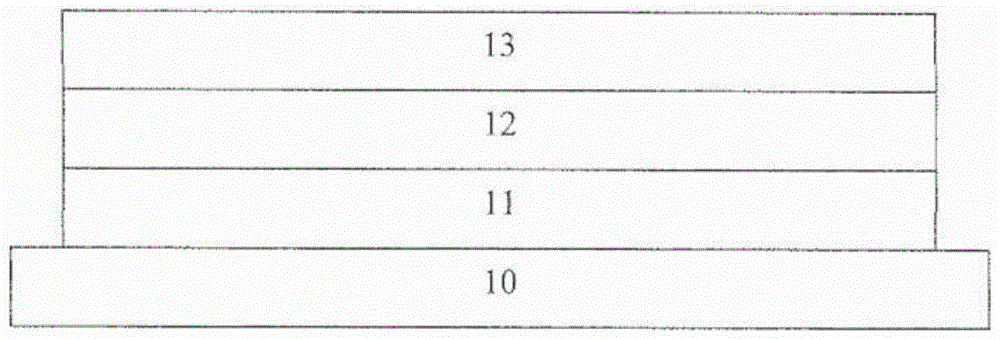
图1是表现形成太阳能电池的层的堆叠体的示意图。
图2是表现包含电子传输层(ETL)的太阳能电池的层的堆叠体的示意图。
图3是器件例2中使用的太阳能电池的示意图。
图4是根据器件例2的OSC的电流–电压特性,所述器件例包含化合物C13作为ETL中的n-掺杂剂。
具体实施方式
根据图1,有机太阳能电池至少包含衬底10、阳极11、光吸收单元12和阴极13。所述层的堆叠体也可以颠倒,其中层11将是阴极,和层13将是阳极。所述有机太阳能电池中可提供附加的光吸收单元。
在一种实施方式中,衬底10可以是透明衬底,例如玻璃、或聚合的板或网。阳极11可以是透明导电氧化物,例如ITO、FTO、AlZO。阴极13可包含铝或铝合金。或者,光吸收单元12可包含供体聚合物、优选含噻吩的聚合物和受体、优选富勒烯或可溶性富勒烯衍生物的掺合物。在这种实施方式中,在所述光吸收单元12和阴极13之间形成包含所述式1化合物(例如掺杂的电子传输层)或由其组成(例如电子提取层)的附加层。任选地,所述层结构可以颠倒。
在备选实施方式中,阳极11是不透明的并且主要包含铝或铝合金。衬底10不一定是透明的。阴极13包含透明导电氧化物层或厚度小于30nm的薄透明金属层。
仍然结合图1,在另一种实施方式中,衬底10、阳极11和阴极13是透明的。在这种实施方式中,整体器件是半透明的,因为它对于可见区波长内任何波长的入射光并不具有100%吸收。
也可以提供多重堆叠的器件(例如串联器件)。在这样的器件中,在光吸收单元12和阴极13之间形成至少一个附加的光吸收单元。可以形成附加的有机或无机层来提供合适的电子连接和光学优化所述层位置。优选地,这些功能的至少部分由包含所述式1化合物的层提供。
图2显示了表现有机太阳能电池的层的堆叠体,所述有机太阳能电池包含衬底20、阳极21、包含吸收层的光吸收单元22、有机电子传输层(ETL)23和阴极24。所述层的堆叠体也可以颠倒。所述ETL可以在阴极24和吸收层22之间形成。所述太阳能电池中可提供附加的光吸收单元。
在一种实施方式中,有机电子传输层23可以包含电子传输基质(ETM)化合物作为它的主要组分和所述式1的化合物作为掺杂剂。所述掺杂的ETL 23可具有任何厚度。在光吸收层22和阴极24之间没有附加吸收层的情况下,它的厚度优选小于50nm。
结合图1描述的所有实施方式也可适用于图2的太阳能电池。
所有图都是太阳能电池的层状结构的示意性表现。一些器件特征没有显示,例如电连接、封装、电极外部的光学结构等等。层厚度不是按比例绘制的。至少一个所述电极(阳极和/或阴极)在所述器件活性的波长范围内是透明的。
在另一种实施方式中,光吸收单元22是供体-受体本体异质结,例如供体-受体材料的掺合物。所述供体优选由包含吡咯或噻吩基团的强吸收化合物形成。所述受体优选是C58、C60或C70富勒烯或可溶性富勒烯衍生物。所述ETL 23可包含所述式1的化合物作为掺杂剂。
在表1中,优选的示例性的式1化合物与如果10wt%的本发明化合物掺杂到ETM E1、E2、E3之一中所达到的电导率一起列出。HOMO值通过循环伏安法在二氯甲烷(DCM)中测量,带有星号的值是在四氢呋喃(THF)中,n.s.是指“没有信号”。
E1代表富勒烯C60(CAS 99685-96-8,LUMO为-1.0V vs.Fc+/Fc,参见Chem.Rev.2000,vol.100,1075页,表1),
E2代表
(CAS 256642-92-9,LUMO为-0.83V vs.Fc+/Fc),
E3代表
(CAS 1415745-03-7,在WO2012/16838中报告,LUMO为-1.01V vs.Fc+/Fc)。
表1
在某些电导率值下面列出并用双星号突出的第二个值给出了最大电导率的温度,℃。如果温度上升到超过这个值,则观察到逐步的电导率降低。在OPV中,如果最大电导率温度高于100℃是有利的。优选它高于110℃,更优选高于120℃,更加优选高于130℃,最优选高于140℃。得到的结果显示,所提供的化合物允许在用于OPV内的典型ETM中有效n-掺杂,并且电导率的温度稳定性很好。
实施例
辅助程序
所述合成用可商购的起始化合物和无水溶剂进行,所述起始化合物和无水溶剂没有另行提纯。13C NMR光谱在氘代氯仿作为溶剂中在125MHz下测量。
循环伏安法
在具体化合物处给出的氧化还原电位在待测物质的氩气脱气的无水0.1M THF溶液中测量,所述测量在氩气氛下,有0.1M四丁基六氟磷酸铵支持电解质,在铂工作电极之间并有由氯化银覆盖的银丝组成并直接浸泡在所述测量溶液中的Ag/AgCl假标准电极,扫描速率为100mV/s。第一次运行在工作电极上最宽的电位设置范围内进行,然后在随后的运行内适当调节所述范围。最后三次运行在添加二茂铁(0.1M浓度)作为标准下进行。所研究的化合物的阴极和阳极峰值所对应的电位的平均值,在减去对标准Fc+/Fc氧化还原耦观察的阴极和阳极电位平均值后,最终提供了上面报告的值。
合成例1:
N-(氯(二甲基氨基)亚甲基)-N-甲基甲铵氯化物(I2)
37.8mL(440mmol)草酰氯在氩气氛下缓慢添加到10.5mL(88mmol)1,1,3,3-四甲基脲在60mL氯仿的溶液中。85℃下(在回流下)搅拌16小时后,蒸馏掉溶剂,残渣用二乙醚洗涤。在真空中干燥后,得到14.9g(87.6mmol;99.5%)N-(氯(二甲基氨基)亚甲基)-N-甲基甲铵氯化物。
合成例2:
2-氯-1,3-二甲基-1H-苯并[d]咪唑-3-鎓六氟磷酸盐(I3)
向含12.5g(81.9mmol)2-氯-1H-苯并[d]咪唑的175ml水添加20.6g(245.7mmol)碳酸氢钠和46.6mL(491.5mmol)硫酸二甲酯。所述混合物在80℃搅拌10小时。冷却到0℃之后,添加30mL六氟磷酸(V)氢。过滤所述沉淀物,用水洗涤并在真空中干燥,产生14.84g(45.44mmol;55%)2-氯-1,3-二甲基-1H-苯并[d]咪唑-3-鎓六氟磷酸盐。
合成例3:
1-(氯(哌啶-1-基)亚甲基)哌啶-1-鎓氯化物(I4)
33mL(38.4mmol)草酰氯在氩气氛下缓慢添加到含15.3g(7.8mmol)二(哌啶-1-基)甲酮的150mL氯仿中。所述混合物在80℃搅拌20小时。在蒸馏掉溶剂并在真空中干燥后,1-(氯(哌啶-1-基)亚甲基)哌啶-1-鎓氯化物没有进一步提纯就用于接下来的合成步骤。
合成例4:
1,4-亚苯基二酰亚胺基光气中间体(I6)
在借助于冰冷却浴保持温度在0和25℃之间的50.00g 1,4-亚苯基二异氰酸酯在250mL氯仿的搅拌过的溶液中,在大约1小时期间内引入干燥气态氯,直到气体吸收停止。在RT下搅拌另外3小时后,旋转蒸发所述溶液,给出71g浅灰色结晶固体,其从700mL EE结晶。得到61.3g白色结晶固体。LC/MS 270(M)。
合成例5:
N1,N2,N4,N5-四(1,3-二甲基咪唑烷-2-亚基)苯-1,2,4,5-四胺(C1)
含1.49g(8.8mmol)2-氯-1,3-二甲基-4,5-二氢-1H-咪唑-3-鎓氯化物(可商购的中间体I1)的24mL乙腈在氩气氛下于0℃添加到0.5g(1.76mmol)苯-1,2,4,5-四胺四盐酸盐在10mL乙腈和3.2mL三乙胺的悬液中。所述混合物在0℃搅拌1.5小时。在过滤所形成的沉淀和蒸馏溶剂后,残渣溶解在盐酸水溶液(10wt%浓度)中并用氢氧化钠水溶液(20wt%)碱化。过滤出沉淀,用水洗涤并在真空中干燥,产生0.92g(1.76mmol;理论收率的100%)白色固体。所述产物通过梯度升华提纯以供分析表征。
熔点:290℃
合成例6:
N3,N3',N4,N4'-四(1,3-二甲基咪唑烷-2-亚基)-[1,1'-联苯基]-3,3',4,4'-四胺(C2)
含2.00g(11.83mmol)2-氯-1,3-二甲基-4,5-二氢-1H-咪唑-3-鎓氯化物(I1)的10mL乙腈在氩气氛下添加到0.61g(2.85mmol)联苯基-3,3',4,4'-四胺在20mL乙腈和4.6mL三乙胺的悬液中。所述混合物在室温下搅拌2天。过滤出沉淀并蒸馏溶剂后,残渣悬浮在2M氢氧化钠溶液中并在45℃搅拌5分钟。过滤后得到1.17g(1.95mmol;68%)灰白色固体,用水洗涤并在真空中干燥。所述产物通过梯度升华提纯以供分析表征。
熔点:231℃
合成例7:
N1-(1,3-二甲基咪唑烷-2-亚基)-N4,N4-双(4-((1,3-二甲基咪唑烷-2-亚基)氨基)苯基)苯-1,4-二胺(C3)
含2.00g(11.9mmol)2-氯-1,3-二甲基-4,5-二氢-1H-咪唑-3-鎓氯化物(I1)的20mL乙腈在氩气氛下添加到1.00g(3.4mmol)N1,N1-双(4-氨基苯基)苯-1,4-二胺在30mL乙腈和3.8mL三乙胺的悬液中。所述混合物在室温下搅拌24小时。蒸馏溶剂后,残渣悬浮在2M氢氧化钠溶液中并在45℃搅拌5分钟。过滤后得到1.4g(2.42mmol;71%)玫瑰色固体,用水和丙酮洗涤并在真空中干燥。所述产物通过梯度升华提纯以供分析表征。
熔点:226℃
合成例8:
2',2”,2”',2”'-(苯-1,2,4,5-四基)四(1,1,3,3-四甲基胍)(C4)
含14.9g(87.6mmol)N-(氯(二甲基氨基)亚甲基)-N-甲基氯化甲铵氯化物(I2)的250mL乙腈在氩气氛下于0℃添加到5g(17.6mmol)苯-1,2,4,5-四胺四盐酸盐在100mL乙腈和51mL三乙胺的悬液中。所述混合物在0℃搅拌2小时。蒸馏溶剂后,残渣溶解在盐酸水溶液(10浓度)中并用20wt%氢氧化钠水溶液碱化。用甲苯提取,用乙腈洗涤并在真空中干燥,产生3.16g(5.96mmol;34%)白色固体。所述产物通过梯度升华提纯以供分析表征。
熔点:206℃
合成例9:
N1,N4-双(1,3-二甲基咪唑烷-2-亚基)-2-甲氧基苯-1,4-二胺(C5)
第一步
3.0g(17.8mmol)2-甲氧基-4-硝基苯胺和0.8g钯炭(10wt%)添加到100ml四氢呋喃(THF)中。小心添加含8.66mL(114mmol)一水合肼的40ml THF并将所述反应混合物在90℃搅拌3小时。冷却之后,过滤所述悬液并用THF洗涤所收集的固体。滤液在减压下缩减成灰色残渣。2.44g(17.66mmol,99%)2-甲氧基苯-1,4-二胺在氩气下储存并且无需进一步提纯即使用。
第二步
含2.00g(11.9mmol)2-氯-1,3-二甲基-4,5-二氢-1H-咪唑-3-鎓氯化物(I1)的20mL乙腈在氩气氛下添加到0.66g(4.7mmol)2-甲氧基苯-1,4-二胺在20mL乙腈和2.4mL三乙胺的悬液中。所述混合物在室温下搅拌50小时。过滤沉淀并蒸馏溶剂后,残渣悬浮在2M氢氧化钠溶液中并在45℃搅拌5分钟。过滤出沉淀,蒸馏掉溶剂,残渣悬浮在乙腈/甲醇混合物中并通过氧化铝垫(Alox N/UV254)过滤。在真空中干燥后得到1.2g(3.63mmol;77%)橙色固体。所述产物通过梯度升华提纯以供分析表征。
熔点:149℃
合成例10:
N1,N4-双(1,3-二甲基-1H-苯并[d]咪唑-2(3H)-亚基)苯-1,4-二胺(C6)
含14.84g(45.44mmol)2-氯-1,3-二甲基-1H-苯并[d]咪唑-3-鎓六氟磷酸盐(I3)的50mL乙腈在氩气氛下于0℃添加到1.97g(18.18mmol)苯-1,4-二胺在250mL乙腈和15.7mL三乙胺的悬液中。所述混合物在室温下搅拌20小时。过滤出沉淀,用乙腈洗涤,悬浮在2M氢氧化钠溶液中并在45℃搅拌5分钟。过滤后得到6.42g(11.65mmol;64%)白色固体,用水洗涤并在真空中干燥。所述产物通过梯度升华提纯以供分析表征。
熔点:290℃
合成例11:
N1,N2,N4,N5-四(1,3-二甲基-1H-苯并[d]咪唑-2(3H)-亚基)苯-1,2,4,5-四胺(C7)
含15.3g(46.84mmol)2-氯-1,3-二甲基-1H-苯并[d]咪唑-3-鎓六氟磷酸盐(I3)的50mL乙腈在氩气氛下添加到2.66g(9.37mmol)苯-1,2,4,5-四胺四盐酸盐在250mL乙腈和16mL三乙胺的悬液中。所述混合物在室温下搅拌20小时。过滤出沉淀,用乙腈洗涤,悬浮在2M氢氧化钠溶液中并在45℃搅拌5分钟。过滤后得到5.7g(7.97mmol;85%)白色固体,用水洗涤并在真空中干燥。所述产物通过梯度升华提纯以供分析表征。
熔点:374℃
合成例12:
N1,N2,N4-三(二(哌啶-1-基)亚甲基)苯-1,2,4-三胺(C8)
含19.6g(78mmol)1-(氯(哌啶-1-基)亚甲基)哌啶-1-鎓氯化物(I4)的150mL乙腈和65mL三乙胺在氩气氛下添加到5.1g(26mmol)苯-1,2,4-三胺二盐酸盐在160mL乙腈和22mL三乙胺的悬液中。所述混合物在室温下搅拌72小时。过滤沉淀并蒸馏掉溶剂。残渣通过在氯仿/甲醇中柱层析和通过从二氯甲烷溶液在己烷中沉淀而提纯,产生2g(3.04mmol;12%)泡沫状固体。所述产物通过梯度升华提纯以供分析表征。
合成例13:
N1,N2,N4-三(1,3-二甲基-1H-苯并[d]咪唑-2(3H)-亚基)苯-1,2,4-三胺(C9)
含11.7g(35.83mmol)2-氯-1,3-二甲基-1H-苯并[d]咪唑-3-鎓六氟磷酸盐(I3)的25mL乙腈在氩气氛下于0℃添加到1.79g(9.13mmol)苯-1,2,4-三胺二盐酸盐在40mL乙腈和12.7mL三乙胺的悬液中。所述混合物在室温下搅拌72小时。过滤出沉淀,用乙腈洗涤,悬浮在2M氢氧化钠溶液中并在45℃搅拌5分钟。过滤后得到2.5g(4.5mmol;49%)灰色固体,用水和乙腈洗涤并在真空中干燥。所述产物通过梯度升华提纯以供分析表征。
合成例14:
N1-(1,3-二甲基-1H-苯并[d]咪唑-2(3H)-亚基)-N4,N4-双(4-((1,3-二甲基-1H-苯并[d]咪唑-2(3H)-亚基)氨基)苯基)苯-1,4-二胺(C10)
含3.15g(9.65mmol)2-氯-1,3-二甲基-1H-苯并[d]咪唑-3-鎓六氟磷酸盐(I3)的25mL乙腈在氩气氛下于0℃添加到0.75g(2.57mmol)N1,N1-双(4-氨基苯基)苯-1,4-二胺在75mL乙腈和3.2mL三乙胺的悬液中。所述混合物在室温下搅拌48小时。过滤沉淀,用乙腈洗涤,悬浮在2M氢氧化钠溶液中并在45℃搅拌5分钟。过滤后得到0.9g(1.25mmol;49%)泡沫状固体,用水洗涤并在真空中干燥。所述产物通过梯度升华提纯以供分析表征。
合成例15:
3,3'-(1,4-亚苯基双(亚氮烷基))双(N1,N1,N2,N2-四甲基环丙-1-烯-1,2-二胺)(C11)
25ml烧瓶装入10mL乙腈、1g(2mmol)氯代双(二甲基氨基)环丙基鎓六氯锑酸盐(可商购的中间体I5)和0.6mL 1.8二氮杂双环[5.4.0]十一-7-烯。添加二氨基苯(86mg,0.8mmol)并将所生成的混合物保持在90℃过夜。冷却之后,过滤所述悬液并用乙腈洗涤深色的滤饼。滤液在真空中浓缩,给出红色油,所述油溶解在二氯甲烷中。有机层用稀氢氧化钠溶液洗涤并用水洗涤两次。用硫酸镁干燥后,在真空中除去溶剂。分离出54mg(19%)淡黄色的固体。
合成例16:
N,N'-双-(二-吗啉-4-基-亚甲基)-苯-1,4-二胺(C12)
8.10g(30mmol,1eq)1,4-亚苯基二酰亚胺基光气中间体(I6)在氮气下溶解在150mL无水THF中,在冰浴中冷却下添加42mL(480mmol,16eq)吗啉(放热),在RT下搅拌16h,白色悬液在旋转蒸发器上真空下蒸发,添加75mL吗啉,在110℃热油浴中加热5h,所述悬液转成棕黄色,在RT再搅拌16h。所述反应混合物溶解在750mL氯仿中,用375mL 2M NaOH水溶液提取,水相用375mL氯仿提取两次,合并的有机相用188mL 2M NaOH和375mL盐水提取,用硫酸钠干燥并旋转蒸发,给出15.5g浅棕色固体。粗产物通过用150mL纯乙醇煮沸而提纯,所述悬液冷却到RT,过滤,用纯乙醇洗涤并在真空中干燥。得到的13.69g白色固体根据NMR探查包含96%纯度与4%溶剂,通过从异丙醇结晶进一步提纯。
元素分析:C 60.79%(理论值61.00),H 7.55%(理论值7.68),N17.64%(理论值17.78)。LC/MS-ESI 473(M+H),13C-NMR:48.80,66.75,122.31,144.46和157.09ppm。
合成例17:
N”-[4-(N',N”-二甲基-N',N”-二苯基胍基)-苯基]-N,N'-二甲基-N,N'-二苯基胍(C13)
5.40g(20mmol,1eq)1,4-亚苯基二酰亚胺基光气中间体(I6)在氮气下与34.6mL纯N-甲基苯胺(320mmol,16eq)混合,在没有放热效应下形成浅绿色的悬液。在加热下,放热反应在60℃开始,所述混合物在90℃下在大约30min期间固化。冷却,用超声浴悬浮在150mL二乙醚中,过滤浅灰色固体,溶解在300mL氯仿中,用160mL 1M NaOH水溶液提取,水相用50mL氯仿提取两次,合并的有机提取物用100mL1M NaOH和100mL盐水洗涤,经硫酸钠干燥,过滤并且滤液在真空下旋转蒸发。得到18.2g浅粉色固体,通过用甲苯以及随后用乙醇和氯仿煮沸提纯,给出最终通过从异丙醇结晶提纯的白色固体。
元素分析:C 77.79%(理论值78.23),H 6.53%(理论值6.57),N 15.11%(理论值15.20)。LC/MS-APCI 552(M),13C-NMR:39.63,117.41,120.15,122.10,123.81,128.51和153.79ppm。
合成例18:
N,N,N',N'-四苯基-N”-[4-(N',N',N”,N”-四苯基-胍基)-苯基]-胍(C14)
5.00g(18.5mmol,1eq)1,4-亚苯基二酰亚胺基光气中间体(I6)在氮气下与通过在70℃暖油浴中加热反应烧瓶熔化的50.143g纯二苯胺(296mmol,16eq)混合。100℃下一小时后,原来的黄绿色均匀混合物转成棕色。冷却到RT,与50mL二乙醚混合,用超声处理,直到油状粘稠混合物转为黄色悬液。添加100mL饱和的NaHCO3水溶液,分离橙褐色有机相,水相用50mL EE提取两次,合并的有机相用50mL饱和NaHCO3水溶液提取并旋转蒸发而形成47g黄色油,所述黄色油添加300mL EE后在超声浴中形成微悬浮液,其过滤和干燥后给出6.26g黄色固体。所述粗产物通过随后从甲苯和异丙醇结晶而进一步提纯。
元素分析:C 83.80%(理论值83.97),H 5.63%(理论值5.54),N10.40%(理论值10.49)。LC/MS-ESI 801(M+H),13C-NMR:121.28,123.88,124.10,124.33,125.00,128.54,128.69,143.22,144.30,144.86,150.50ppm。
合成例19:
N”-[4-(N',N',N”,N”-四-对甲苯基胍基)-苯基]-N,N,N',N'-四-对甲苯基胍(C15)
5.24g(20.0mmol,1eq)1,4-亚苯基二酰亚胺基光气中间体(I6)在氮气下与通过85℃暖油浴加热反应烧瓶熔化的63.13g纯p,p'-二甲苯胺(320mmol,16eq)混合。在100℃三小时后,所述棕色溶液已经冷却,用500mL氯仿稀释并与250mL 2M NaOH水溶液一起搅拌。分离有机相,水相用250mL氯仿提取两次,合并的有机相随后用250mL 2M NaOH、250mL盐水提取,经硫酸钠干燥并旋转蒸发,给出70g粘稠的物质,所述物质溶解在300mL EE中,浓缩,溶解在200mL沸腾乙醇中,并通过冷却到RT结晶。得到的粗产物在二氧化硅柱上用EE:石油醚作为洗脱液层析,给出6.97g黄色结晶固体。13C-NMR:20.84,20.92,121.17,122.53,123.85,124.70,128.35,128.93,129.14,132.29,132.96,133.52,139.73,142.08,142.60,143.15,143.30,146.02,150.79ppm。
在器件例中,使用下列辅助化合物:
N4,N4,N4”,N4”-四([1,1'-联苯基]-4-基)-[1,1':4',1”-三联苯基]-4,4”-二胺(HT1,CAS 925431-34-4)作为空穴传输基质,1,2,3-三亚基三(氰基亚甲烷基))三-(2,3,5,6-四氟苄腈)-环丙烷(PD2,CAS1224447-88-4),四(1,3,4,6,7,8-六氢-2H–嘧啶并[1,2-a]嘧啶根合)二钨(II)(W2(hpp)4,CAS 463931-34-2)
器件例1
pn结器件用于评估所述式1的新掺杂剂与强供体W2(hpp)4。利用ITO作为阳极、由空穴传输基质HT1和p-掺杂剂PD2以重量比9:1组成的50nm p-掺杂HTL(空穴传输层)、由富勒烯C60作为基质、用所述式1的新掺杂剂之一以重量比7:3掺杂组成的50nm电子传输层、和Al阴极,在玻璃衬底上制作所述pn-结器件。5mA/cm2的电流密度所必需的电压对于化合物C7为0.02V,对于化合物C12为0.64V,对于化合物C13为0.65V,对于化合物C14为0.30V和对于化合物C15为0.78V。化合物C7的值好得出人意料,与W2(hpp)4(HOMO<<-1.0V vs.Fc+/Fc)相比给出了低得多的供给强度,它们在相同的排列中允许对于5mA/cm2的电流密度在0.01V的电压下操作所述pn结。化合物C12-C15,根据表1中列出的它们的氧化还原电位具有甚至更低的还原强度,仍然允许在低于1V下操作所述pn结。
器件例2
为了评估本发明的亚胺化合物在半导体材料中的适用性而设计的试验光伏器件的层结构如图3示意性显示。所述器件在带有90nm ITO阴极31的玻璃衬底30上通过真空沉积下列层而制备:由富勒烯C60以9:1的重量比用式1化合物掺杂制成的10nm厚的ETL 32,由C60制成的20nm厚的吸收层33,由C60和锌酞菁以重量比1:1制成的30nm厚的吸收层34,由HT1制成的2nm厚的空穴提取层35,由HT1以重量比19:1用PD2掺杂制成的40nm厚的HTL 36,由纯PD2制成的2nm厚的空穴注入层37,和100nm厚的铝阴极38。对于几种本发明的电子传输材料的器件性能的比较在表2提供。
表2
得到的结果意外地显示,即使具有少于-0.3V vs.Fc+/Fc的负氧化还原电位的式1化合物仍能成功地在用于OSC的半导体材料中使用。
以上说明书、权利要求书和附图中公开的本发明的特征个别以及以任何组合都可以是对于以各种实施方式实现本发明而言重要的。
在整个申请中使用的缩写
AlZO 氧化铝锌
APCI 大气压化学电离
CAS 化学文摘服务社(Chemical Abstract Service)参考编号
CV 循环伏安法
DCM 二氯甲烷
EE 二乙醚
EIL 电子注入/提取层
ESI 电喷雾电离
ETL 电子传输层
ETM 电子传输基质
Fc 二茂铁
Fc+ 二茂铁鎓
FTO 氟掺杂的氧化锡
HBL 空穴阻挡层
HIL 空穴注入层
HOMO 最高占有分子轨道
HPLC 高效液相色谱
HTL 空穴传输层
HTM 空穴传输基质
ITO 氧化铟锡
LC 液相色谱
LUMO 最低未占分子轨道
mol.% 摩尔百分比
MS 质谱
NMR 核磁共振
OLED 有机发光二极管
OPV 有机光伏器件
OSC 有机太阳能电池
OVPD 有机气相沉积
QE 量子效率
PCBM 苯基C61丁酸甲酯
Rf TLC中的阻滞因数
TCNQ 四氰基醌二甲烷
Tg 玻璃化转变温度
TLC 薄层色谱
vs 对比
VTE 真空热蒸发
wt% 重量百分比
- 还没有人留言评论。精彩留言会获得点赞!