碳点-铂-钯复合体的制备方法、由此制备的碳点-铂-钯催化剂及利用其的燃料电池与流程
本发明涉及均匀分散有铂催化剂,表面积宽且稳定,催化剂活性高的铂系纳米复合体的制备方法和由此制备的燃料电池用铂系催化剂及利用其的燃料电池。
背景技术:
燃料电池作为使燃料的化学能通过电化学反应直接转换成电能及热能的装置,被考虑为对于新能量要求的解决方案。在燃料电池中,直接甲醇型燃料电池(dmfcs,directmethanolfuelcells)和直接甲酸型燃料电池(dfafcs,directformicacidfuelcells)作为便携式电子设备和无污染汽车的重要部件深受瞩目。直接甲醇型燃料电池和直接甲酸型燃料电池的最大的优点为具有高效率性、高能量密度、低的工作温度、低的毒性,可在确保碳化氢燃烧时产生的副产物之类的污染物质不产生的情况下生产电力。但因对于氧化反应的活性低,作为氧化电极(anode,阳极)催化剂的铂系催化剂的价格昂贵的缺点而在更多范围内的应用受限。
铂(pt)系催化剂为在碱性电解质溶液中对甲醇及甲酸的氧化最有希望的催化剂。但是,铂系催化剂因吸附于表面的co和/或反应中间体而容易被中毒(poisioning)。最终,铂系催化剂的电子催化剂活性随着反应时间的经过甚至在数百秒内快速降低。为了增加催化剂的耐中毒性,提高活性来提高电子催化剂性能,并减少催化剂中昂贵的铂的使用量,正在开发如pt-ru、pt-pd、pt-co、pt-ti、pt-v及pt-au等作为铂和其他金属的合金形态的二元催化剂(韩国授权专利10-1137066,美国授权专利8603400),已被商用化。并且,也正在对三元类催化剂、四元类催化剂进行研究。
与一般的固体催化剂相比,就中空性(hollow)铂系纳米粒子而言,表面积宽,密度低,且对于甲醇和甲酸的氧化的电子催化剂性能高。中空性铂系纳米粒子的电子催化剂性能可通过调节纳米粒子的形状、大小及组成来得到进一步的提高。为此,作为铂系催化剂,提出了制备中空性纳米粒子的很多方法,据报告,有如pt-co、pt-ni、pt-pd等形状和组成不同的多种基于铂的中空性纳米粒子。尽管如此,就多种上述中空性纳米催化剂而言,其合成方法复杂,结构致密,且表面光滑,因而仍存在表面活性不高的问题。
另一方面,开发了如下的多种催化剂,为了减少多种铂系催化剂的凝聚,并增加电导率来使通过反应产生的电流有效流动,使铂系催化剂浸涂于碳黑、碳纳米管、石墨烯、石墨烯氧化物的还原物、无定形碳之类的碳纳米物质的载体中(韩国授权专利10-0480969)。但是,就浸涂于碳载体的催化剂而言,在燃料电池工作期间,多种催化剂金属纳米粒子从碳载体分离,而这成为大大减少催化剂的有效表面积来降低燃料电池性能的主要原因。进而,多种碳载体具有与多种其他物质相结合的性质,当制备工作电极时,夹在很多纳米晶体之间,致使有效表面积大大减少。为了克服这种多种碳载体物质的缺点,作为碳纳米物质的新形态,开发了碳点(cdot,carbondot)。
碳点作为具有独特性质的新型碳原材料,在光电子、能量储存及转换设备、太阳能电池、生物传感器、生物技术中被认定其潜在的适用可能性,从而深受瞩目。碳点作为含氧的准球形(quasi-sphericalshape)的碳质纳米粒子,其大小为10nm以下。碳点的表面包含大量的含氧的官能团,因而亲水性高,且可均作为电子供体及电子受体起作用。碳点还可利用为与抗坏血酸之类的还原剂相结合来在水溶液中使金属纳米粒子稳定的结构体。碳点和金属纳米粒子的融合可在新的杂交体(hybrid)中引起固有的性质变化,以提高催化剂、电、光学性能。碳点不仅进行生物合成,而且具有高的电导率特性、很大的表面积、低的毒性及化学稳定性,因而作为燃料电池领域的电极/电极载体以有希望的原材料备受瞩目。但是,对于碳点/金属复合体,至今,只进行极其受限的研究,已报告的碳点/金属复合体并非是多孔性结构,因而其表面活性区域少,且催化剂活性也低(guo等,acscatalysis2015,5,2903)。
技术实现要素:
技术问题
本发明的目的在于,提供均匀分散有铂催化剂,表面积宽且稳定,对于co等的耐中毒性和催化剂活性高的铂系纳米复合体,以解决如上所述的现有技术的问题。
并且,本发明的目的在于,提供可通过简单的方法制备上述铂系纳米复合体的合成方法。
本发明的另一目的在于,提供包含上述纳米复合体的燃料电池用催化剂及包含上述催化剂的燃料电池。
技术方案
用于实现上述目的的本发明涉及铂、钯及碳点的纳米复合体(以下,称为“cdot/ptpd纳米复合体”)的制备方法,其特征在于,包括:步骤(a),在混合碳点和铂前体及钯前体的状态下,使铂前体及钯前体还原来制备致密的纳米树枝状球形纳米复合体;以及步骤(b),在上述致密的纳米树枝状球形纳米复合体中选择性地蚀刻钯来赋予多孔性。
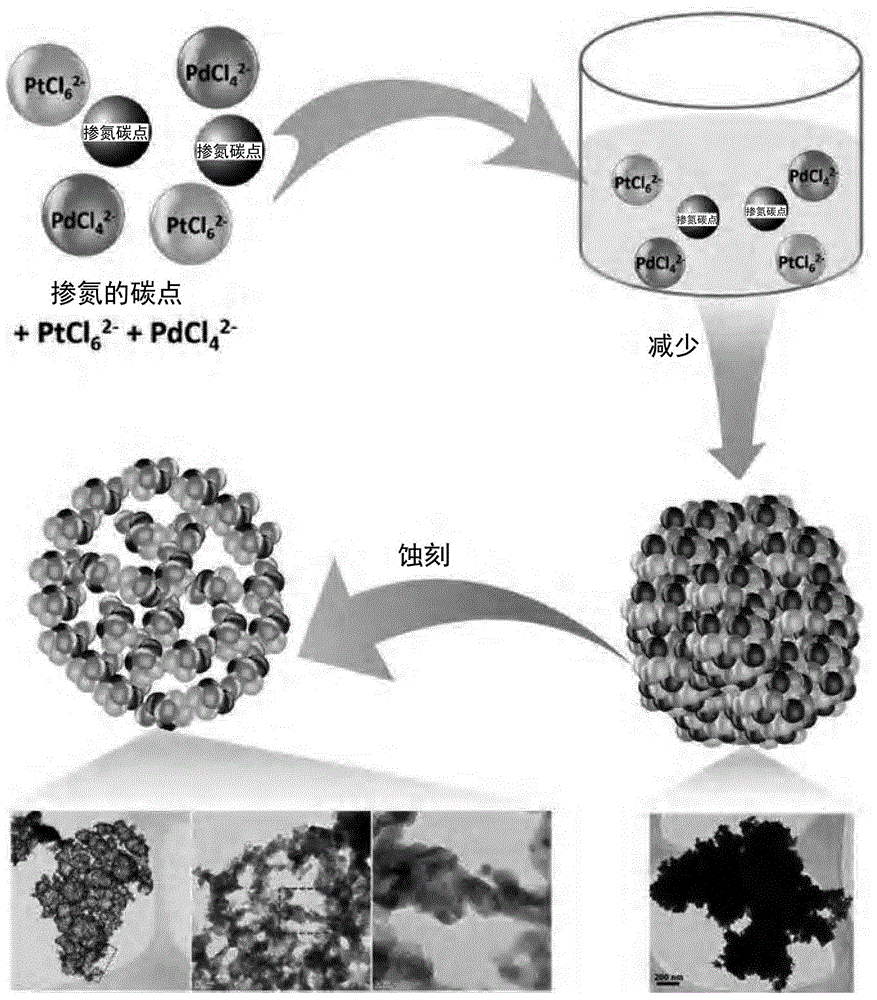
图1为本发明的cdot/ptpd纳米复合体的制备方法的示意图。参照图1,对本发明的cdot/ptpd纳米复合体的制备方法进行详细说明。
本发明的cdot/ptpd纳米复合体通过两个步骤的反应来制备而得。
(a)致密的纳米树枝状球(densenanodendriticsphere)形纳米复合体的制备
第一个步骤为在混合碳点(以下,称为“cdot”)和铂前体及钯前体的状态下,使铂前体及钯前体还原来制备致密的纳米树枝状球形纳米复合体的步骤。
上述碳点在铂前体和钯前体还原时起到生长核作用,以形成纳米树枝状的球。在碳点不存在的状态下,若使铂前体和钯前体还原,则形成有凝聚了表面光滑的ptpd合金的粒子,以代替纳米树枝状的球,且平均粒径也为400~600nm,形成有相比于碳点存在的状态下制备的粒子大2~4倍程度的粒子。
据报告,上述碳点在现有技术中有多种制备方法(wangy.,hua.carbonquantumdots:synthesis,propertiesandappliations.j.mater.chem.c.,2014;2:6921;qus,wangx,luq,liuxandwangl.abiocompatiblefluorescentinkbasedonwater-solubleluminescentcarbonnanodots.angewcheminted2012;51:12215),本发明不涉及碳点本身的制备方法,因而碳点的制备方法可使用任意方法。并且,更优选地,在上述碳点中还掺氮。氮具有不仅改善表面的亲水性,还提高作为电子供体和电子受体的特性,且容易导入官能团等改善碳点的特性的效果。对此,在以下实施例中,作为代表例,记载了掺氮的碳点的具体结果,但可确认到当使用未掺杂在实施例1的第一步骤中制备的氮的碳点时,也同样在铂前体和钯前体还原时起到生长核作用,以形成纳米树枝状的球。众所周知,掺氮的碳点的制备方法也同样在现有技术中有多种方法,因而只要是本发明所属技术领域的普通技术人员(reviewonrecentadvancesinnitrogen-dopedcarbons:preparationsandapplicationsinsupercapacitors,journalofmaterialschemistrya,2016,4,1144-1173),就可以利用其来容易进行制备。
当使用n-碳点时,相对于碳,氮的掺杂量优选为20重量百分比以下。当氮的掺杂量过多时,不仅难以形成掺氮的碳点,在铂前体和钯前体还原时作为生长核的功效也降低。
碳点的大小优选为1~10nm,但存在当碳点的大小过大时,难以形成纳米树枝状球的问题。当掺氮的碳点的大小过小时,难以合成碳点。
上述铂前体包含铂离子,只要可以通过还原反应来还原为铂,就可以使用任何一种。作为铂前体的具体例,可使用四氯铂酸(h2ptcl4)、六氯铂酸(h2ptcl6)、四氯铂酸钾(k2ptcl4)、六氯铂酸钾(k2ptcl6)、氯化铂(ptcl2)、四氯化铂(ptcl4)或它们的混合物,但不局限于此。
上述钯前体也包含钯离子,只要可以通过还原反应来还原为钯,就可以使用任何一种。作为钯前体的例,可使用四氯钯酸钠(na2pdcl4)、四氯钯酸钾(k2pdcl4)、六氯钯酸钾(k2pdcl6)、四氯钯酸铵((nh4)2pdcl4)、六氯钯酸铵((nh4)2pdcl6)、氯化钯(pdcl2)或它们的混合物,这也不局限于此。
在上述铂前体、钯前体及碳点的混合物中,当使铂前体、钯前体及碳点的重量为100时,铂前体优选为20~70重量百分比,钯前体优选为70~20重量百分比,碳点优选为3~20重量百分比。当碳点的含量过少时,不足以起到生长核作用,从而难以形成纳米树枝状球,当碳点过多时,催化剂活性降低。当铂前体的比率过少或钯前体的比率过多时,催化剂活性也降低,当铂前体的比率过高或钯前体的含量过少时,不仅催化剂的价格上升,而且在以下步骤中,当选择性地蚀刻钯时,多孔性也受限制。
上述铂前体及钯前体的还原可使用以往制备铂/钯二元金属合金时所使用的任何一种方法。即,可单纯地通过加热反应来实现,或进一步投入还原剂来实现。作为还原剂,可使用选自由甲酸、抗坏血酸、柠檬酸、油胺、甲醛、硼氢化钠、2-甲基-2-吡咯烷组成的组中的一种或两种以上的混合物,并不局限于此。还原反应时的温度为20~180℃,可根据是否使用还原剂,或根据使用的还原剂的种类来选择适当条件。
制备的cdot/ptpd纳米复合体呈致密的纳米树枝状的球形状,表面形成有枝条形状和/或尖的突起。
(b)多孔性赋予步骤
第二个步骤为在第一个步骤中生成的致密的纳米树枝状球形cdot/ptpd纳米复合体中选择性地蚀刻钯来赋予多孔性的步骤。当利用选择性蚀刻剂来处理在步骤(a)中制备的cdot/ptpd纳米复合体时,钯通过脱合金工序来选择性地蚀刻而被去除,接着,发生化学电位差引起的电置换反应(galvanicreplacement)。以下反应式是指示出作为还原剂使用fe3+的情况的脱合金过程的半反应。
阳极(anodic):
阴极(cathodic):
作为上述选择性蚀刻剂,可例举硝酸、feno3或fecl3之类的fe(iii)的盐、h2ptcl6或k2ptcl6之类的pt(iv)盐,只要可以对铂选择性地蚀刻钯,其种类不受限制。利用选择性蚀刻剂来发生反应时的温度为10~300℃,可根据使用的蚀刻剂的种类和浓度、cdot/ptpd纳米树枝状球形复合体中的钯的含量来适当地进行选择。例如,当将fecl3溶液用作选择性蚀刻剂时,若在常温条件下经过5天,则纳米复合体的组成和形态不再有变化,从而可以确认到达到平衡。当将fecl3溶液用作选择性蚀刻剂时,优选地,上述过程在10~50℃温度下进行0.3~10天。可通过调节温度和时间来调节纳米复合体的组成和形态。并且,当然,达到平衡的时间也根据温度而不同。
根据上述选择性蚀刻的结果,在蚀刻初期,在致密的纳米树枝状球的表面开始形成空隙,通过电置换反应随着蚀刻时间的经过,转换成中空性结构,接着,改变为纳米网络结构。因此,可通过调节蚀刻时间来调节纳米复合体的组成和形态。均匀分布于致密的纳米树枝状球形cdot/ptpd纳米复合体内的碳点在以下方面负责重要的作用:在本步骤的脱合金过程中起到骨架结构作用,而且,限制铂和钯的扩散来延迟扩散工序,并防止蚀刻剂渗透到纳米球的下部层,最终,形成中空性结构和/或纳米网络结构。
本发明还涉及一种cdot/ptpd纳米复合体,其特征在于,具有多孔性纳米树枝状的球形状。
在本发明的具有多孔性纳米树枝状的球形状的cdot/ptpd纳米复合体中,碳点和钯均匀分布于整个纳米复合体,相反,铂的表面以更高于铂的内部的浓度存在。因此,当起到催化剂作用时,可提供更多的活性部位。
上述纳米复合体中铂和钯的原子比为76∶24~95∶5,可根据初期铂和钯的组成及选择性蚀刻的条件来进行调节。确认到在选择性蚀刻过程中碳点也受损,ptpd∶c的比率为40∶60~99∶1。初期碳点的浓度越低,越进行蚀刻,碳点的含量越减少。
上述“多孔性”可以为在表面包含多个空隙,或是包含空隙的中空型结构,或多孔性被极大化的纳米网络结构。双重纳米网络结构呈现宽的表面积,其应用性宽,因而最优选。
本发明还涉及包含具有上述多孔性纳米树枝状的球形状的n-cdot/ptpd纳米复合体的燃料电池用催化剂。本发明的燃料电池用催化剂不仅是pt/c或ptpd/c,而且,与根据现有技术的其他燃料电池用催化剂相比,最大电流密度更高,耐中毒性也优秀。可从两个方面说明这种本发明的燃料电池用催化剂的性能提高。首先,因结构上的特征,纳米网络结构的球形结构提供更多的活性区域,因而电子催化剂活性变高。第二,随着ptpd纳米合金结构利用掺氮的碳点来相连接,通过将晶界中电子的移动加速化来增加催化剂活性。
有益效果
如上所述,就本发明的具有多孔性纳米树枝状的球形状的n-cdot/ptpd纳米复合体而言,耐中毒性优秀,因多孔性结构而表面积宽,因而表面活性高,因纳米复合体内的掺氮的碳点的电导率而使电子的移动加速化,因此,可有用地利用为具有高的催化剂活性的燃料电池用催化剂。
并且,就本发明的n-cdot/ptpd纳米复合体而言,制备方法简单,可通过调节合成时的反应条件来容易调节纳米复合体的组成和形态,从而可经济地制备适合要求的组成和形态的纳米复合体。
附图说明
图1为本发明的cdot/ptpd纳米复合体的制备方法的示意图。
图2为本发明一实施例的n-cdot/ptpd纳米复合体的扫描电子显微镜(sem)及透射电子显微镜(tem)图像。
图3为致密的纳米树枝状球形纳米复合体的透射电子显微镜、高分辨率透射电子显微镜(hr-tem)及能量色散x射线光谱仪(edx)元素映射图像。
图4为根据比较例的ptpd合金的扫描电子显微镜及透射电子显微镜图像。
图5为本发明一实施例的n-cdot/pt84pd16纳米复合体的透射电子显微镜、高分辨率透射电子显微镜及能量色散x射线光谱仪元素映射图像。
图6为示出本发明一实施例的n-cdot/ptpd纳米复合体的x射线衍射(xrd)衍射图案的光谱。
图7为本发明一实施例的n-cdot/ptpd纳米复合体的x射线光电子能谱(xps)光谱。
图8为本发明一实施例的n-cdot/ptpd纳米复合体的循环伏安图。
图9为本发明一实施例的n-cdot/ptpd纳米复合体的甲醇及甲酸氧化反应的循环伏安图和示出氧化电流密度的曲线图。
图10为根据比较例的ptpd/c和本发明一实施例的n-cdot/pt84pd16纳米复合体的甲醇及甲酸氧化反应的循环伏安图。
图11为本发明一实施例的n-cdot/ptpd纳米复合体的甲醇及甲酸氧化反应的线性扫描伏安图。
图12为示出本发明一实施例的n-cdot/ptpd纳米复合体的甲醇及甲酸氧化反应的最大电流密度与时间的关系的曲线图。
图13为示出本发明一实施例的n-cdot/ptpd纳米复合体的稳定性的循环伏安图。
图14为示出本发明一实施例的n-cdot/ptpd纳米复合体的甲醇氧化反应的稳定性的循环伏安图。
具体实施方式
以下,列举附图和预先实验及实施例,更详细说明本发明。但是,这种附图和实施例是用于说明本发明的技术思想的内容和范围而例示的,本发明的技术范围并不局限于此或者变更。基于这种例示,在本发明的技术思想的范围内可进行多种变形和变更,这对于本发明所属技术领域的普通技术人员来说是显而易见的。
实施例
实施例1:碳点-铂-钯复合体的制备
1)第一步骤:碳点的制备
(1)掺氮碳点的制备
根据德国应用化学(angewandtechemieinternationaledition)2013,52,7800中记载的方法来制备掺氮碳点(n-cdot)。简单来说,将4g的柠檬酸(citricacidmohohydrate)和1.24g的甘氨酸溶解于10ml的蒸馏水中。在70℃温度下,将溶液处理12小时来使溶剂蒸发,并将剩余的残渣放入高压灭菌器中,以4.3℃/分钟的速度进行升温,在200℃温度下进行3小时水热反应。使用1.0m的naoh溶液来将黑色糖浆状态的产物进行中和,并利用100ml的蒸馏水来稀释。通过透析膜(5kdamwco)对二次蒸馏水透析稀释溶液。对透析液进行冷冻干燥来获得黄色的掺氮碳点。
根据利用透射电子显微镜来观测制备的掺氮碳点的结果,上述掺氮碳点呈直径为1.5~2.5nm的球形。根据高分辨率透射电子显微镜图像,在整个粒子,石墨{100}面以约0.21nm的间隔连续形成有格子边缘(latticefringe)。掺氮碳点溶液呈黄色,当照射365nm的紫外线时,呈现约500nm的最大发光波长,从而以蓝色进行发光。在制备的掺氮碳点的x射线光电子能谱光谱上,缘自c1s、n1s、o1s的峰值分别在286.44、399.82及533.09ev被观测到。从x射线光电子能谱光谱计算出来的c、n、o的比率分别为84.35、5.29及9.36原子百分比。在掺氮碳点的x射线衍射光谱观测到以2θ=20°为主的宽的峰值,这缘自石墨烯结构。在傅里叶变换红外光谱(ft-ir)上,观测到相当于-ch2-、-oh、-nh2、-nh3+、c=o、n-h、c-n、酰胺(amide)的振动的峰值,从而可确认到氮成功地掺杂在碳点中,包含氧和氮的官能团大量存在。
(2)碳点的制备
根据碳(carbon)2016,96,139e144中记载的方法使用微波来制备碳点(cdot)。简单来说,在10ml的蒸馏水中放入3g的柠檬酸(duksan化学)和3g的尿素(shinyochemical)并进行激烈搅拌之后,在微波烘箱(700w)中处理4分钟。最终,在常温条件下,对获得的深棕色溶液进行冷却之后,以7000rpm离心分离30分钟而去除黑色大凝聚体(aggregate)。包含碳点的上清液利用重碳酸盐水来中和,并利用去离子(di)水洗涤多次。回收黑色的碳点之后,在真空烘箱中进行干燥。
将干燥的碳点分散于乙醇中来滴入云母基板(micasubstrate),制备试样之后,使用multimod8(布鲁克(bruker))显微镜,以轻敲模式获得原子力显微镜(afm)图像。根据观测结果,可确认到制备的碳点的平均粒径为1.2nm,呈表示高斯分布的球形(未图示详细数据)。
2)第二步骤:n-cdot/ptpd纳米树枝状球形复合体的制备
在50ml的小瓶中投入将1)中制备的2mg的掺氮碳点分散于20ml的蒸馏水来获得的溶液、20mg的六氯铂酸(h2ptcl6,西格玛奥德里奇(sigma-aldrich))及20mg的四氯钯酸钾(k2pdcl4,西格玛奥德里奇)。在反应混合物中加入1.5ml的甲酸(88%的美国化学会试剂(acsreagent)),盖上小瓶的盖之后,超声波处理5分钟。将均匀混合的溶液放入烘箱,在不搅拌的状态下,在30℃温度下放置24小时。之后,在7000rpm条件下进行离心分离来获得黑色沉淀物,利用乙醇来洗涤多次之后,在70℃温度下干燥24小时。
3)第三步骤:n-cdot/ptpd纳米网络结构的球形复合体的制备
将30mg的fecl3、0.18ml的浓盐酸和7ml的蒸馏水放入20ml的小瓶,搅拌30分钟。在混合液中放入0.7mg的2)中制备的n-cdot/ptpd纳米树枝状球形复合体,在常温条件下放置5天。5天后,在7000rpm条件下进行离心分离来回收黑色沉淀物,依次利用蒸馏水来洗涤三次,利用乙醇来洗涤两次。之后,在70℃的真空烘箱中,干燥24小时之后使用。
比较例:ptpd/c的制备
除了不添加掺氮碳点之外,通过与实施例1的第二步骤相同的方法来反应12小时,制备ptpd纳米合金。之后,将5mg的制备的ptpd纳米合金与20mg的superp碳黑(supperpcarbonblack)进行混合,并命名为ptpd/c。为了以下实施例中的使用,将6mg的ptpd/c分散于1m1的水/ipa/nafion离聚物(ionomer)(iompower,liquion1100)79∶6∶20(v/v)混合溶液来制备油墨。
实施例2:n-cdot/ptpd复合体的物理化学特性的分析
在实施例1中制备的n-cdot/ptpd复合体的形态特性利用场辐射扫描电子显微镜(fe-sem,jsm-6330f)和300kv加速电压的透射电子显微镜(tem,jem-2010hr)进行分析。图2的a)~d)为基于上述分析的扫描电子显微镜图像,e)~h)为透射电子显微镜图像,示出随着n-cdot/ptpd纳米树枝状球形复合体(a,e)的蚀刻时间经过1天(b,f)、3天(c,g)及5天(d,h)获得的n-cdot/ptpd的形态特性。图2的a)为在实施例1的第二步骤中制备的n-cdot/ptpd纳米树枝状球形复合体的扫描电子显微镜图像,示出多个致密的纳米球如项链一样相连接。纳米球的大小为178~195nm,平均直径为186±0.5nm。a)的内部图像为放大图,在表面形成有枝条形状和/或尖的突起而具有纳米树枝状形态。e)的透射电子显微镜图像也示出包含致密的纳米树枝状球的项链形状的纳米结构。
图3示出利用透射电子显微镜(a)、高分辨率透射电子显微镜(hr-tem,300kv,jem-2100f)(b)、高角度环形暗场(haadf)-扫描透射电子显微镜(stem)(c)来观测n-cdot/ptpd纳米树枝状球形复合体的结构的图像和能量色散x射线光谱仪元素映射图像(e~g)。如图3所示,可见在实施例1的第二步骤中制备的n-cdot/ptpd复合体由表面粗糙的致密的纳米球相连接来呈树枝状形态。此时,利用虚线来将纳米球之间的连接点(junction)示于图3的a)中。纳米球中的格子之间间隔为0.237nm,相当于二元ptpd合金的{111}面。能量色散x射线光谱仪元素映射图像中,碳和钯整体上均匀分散于粒子,相反,铂以相对高的浓度存在于粒子的表面。利用电感耦合等离子体质谱(icp-ms,inductivelycoupledplasmamassspectroscopy)(中国上海辰华公司(chenhuaco.,shanghai,china))来分析cdot/ptpd纳米合金融合体的组成的结果显示,铂和钯的元素比为53∶47,在第二步骤中制备的n-cdot/ptpd复合体为致密的纳米树枝状球形的n-cdot/pt53pd47。
图4为在比较例中制备的ptpd合金的扫描电子显微镜(a)及透射电子显微镜(b)图像,示出表面光滑的多个ptpd纳米粒子凝聚的形态而不是纳米树枝状的结构,平均粒径为400~600nm。这启示掺氮碳点在形成树枝状球时起到重要的作用。当掺氮碳点存在时,还原ptpd需要24小时,相反,当掺氮碳点不存在时,经过12小时还原反应才会结束,鉴于此可视为如下,掺氮碳点起到晶核作用,存在于掺氮碳点的表面的多种官能团(例如,-oh、c=o、c-n、c-o-c等)影响pt和pd的还原速度,由此以纳米树枝状球的形态生长。在图2的a)和e)中相互不连接而分离的ptpd合金不存在,这也说明上述观点。
n-cdot/ptpd纳米树枝状球形复合体的脱合金通过从fecl3溶液中浸出(leaching)ptpd合金来实现。随着fecl3溶液的处理时间变长,浸出液从亮黄色逐渐变成棕色,颜色变深,在浸出液的紫外线(uv)光谱中,基于pd2+离子的300nm附近的吸光度逐渐增加。这意味着随着fecl3溶液的处理,通过脱合金从ptpd合金中浸出钯。
根据基于图2的蚀刻时间的n-cdot/ptpd复合体的扫描电子显微镜/透射电子显微镜图像,示出致密的纳米树枝状的球形状的n-cdot/ptpd复合体在蚀刻1天之后在纳米树枝状球形成有空隙(b,f)。纳米树枝状球的平均直径为与蚀刻之前相同的186±0.5nm。根据电感耦合等离子体质谱分析,钯和铂的比率为pt53pd47到pt76pd24,钯的比率减少。
当进一步蚀刻而到蚀刻第三天时,纳米树枝状球凹陷(由黄色箭头表示)而形成中空(图2的c,g),钯和铂的比率也变为pt81pd19。
蚀刻第五天时,钯和铂的比率成为pt84pd16,即使在蚀刻时间进一步经过的7天后,组成也不再有变化,可见浸出反应已结束。图2的d和h示出不仅中空变得更多,而且具有多孔性结构。
图5为示出更详细地观测最终获得的n-cdot/pt84pd16纳米复合体的特性的结果的图像及曲线图。图5的透射电子显微镜图像(a)和高分辨率透射电子显微镜图像(b,c)示出在复合体内多个纳米线相连接而形成具有网络结构的球。格子之间的间隔为0.237nm和0.195nm,分别相当于ptpd合金的{111}和{200}面。相当于高角度环形暗场-扫描透射电子显微镜图像(d)的能量色散x射线光谱仪元素映射(e~g)示出碳和钯均匀分布于球内,铂更多地集中于球的边界。图5的k为示出根据在(i)的高角度环形暗场-扫描透射电子显微镜图像中由箭头表示的纳米线的直径的各个元素的分布的曲线图,在外侧部分,铂的比率也比内侧高,由此可知在表面形成富含铂的结构。
进一步通过x射线衍射图案来分析蚀刻时间对n-cdot/ptpd纳米复合体的结构产生的影响。x射线衍射图案利用使用40kv、100ma的cukα源辐射(sourceradiation)的d2相位系统(phasesystem)(bruker)来获得,图6示出由此获得的x射线衍射光谱。在图6中,24°附近的衍射峰值表示掺氮碳点的{002}碳格子。在蚀刻之间的纳米树枝状球复合体中,(0天)约40.25°、46.8°、68.2°及81.7°的衍射峰值为面心立方(fcc)结晶性ptpd的特征,分别相当于{111}、{200}、{220}和{311}晶面。图6的右侧为放大{111}格子面的衍射峰值的图。随着蚀刻时间的经过,相应的衍射峰值与蚀刻之前相比,逐渐向左侧移动,这与前述的电感耦合等离子体质谱分析结果相同,启示合金的组成中铂的含量增加。
进一步而言,为了获得根据蚀刻时间的表面的元素组成和化学状态的信息,通过作为用于激发的x射线源使用alkαx射线辐射(x-rayradiation)的escalab-mkii光谱仪(spectrometer)(vgco.,英国(unitedkingdom))来测定x射线光电子能谱。图7的a)~c)分别为对于铂的4f、钯的3d及碳的1s区域的x射线光电子能谱光谱。为了查明催化剂的特定原子的状态,利用两对双合透镜来对铂的4f和钯的3d峰值进行反卷积。将电子的结合能和各个氧化状态的相对量整理于下列表1中。
表1
如对于铂4f的图7的a)和表1中可确认,在蚀刻之前的催化剂中,相当于铂金属的4f7/2和4f5/2的强峰值在71.09ev和74.48ev被观测到,相当于pto或pt(oh)2的pt2+的弱峰值在72.11ev和76.48ev被观测到。这暗示随着蚀刻,铂金属4f7/2的结合能与蚀刻之前相比,逐渐弱弱地反向移动,致使铂的电子结构因钯原子而发生变形。尤其,在蚀刻第五天的复合体中,铂金属4f7/2的结合能为70.86ev,向最低的能量移动,这认为这是在n-cdot/pt84pd16复合体中ptpd合金表面结构被最佳化而引起的。
图7的b)涉及钯3d,在蚀刻之前,缘自pd°的3d5/2和3d3/2的峰值在335.55ev及340.8ev被观测到,在相当于pdo的pt2+的337.01ev及343.3ev被观测到。与铂相同,钯金属3d5/2的结合能随着蚀刻时间的经过,逐渐反向移动,移动程度比铂更明显。从x射线光电子能谱光谱计算的纳米网络结构的球形复合体(蚀刻5天后)表面的pt∶pd原子比为91∶9,与利用电感耦合等离子体质谱来测定的大体积复合体的84∶16有差异。这种差异与能量色散x射线光谱仪映射的结果相一致,意味着铂大量分布在纳米复合体的表面。
如表示对于碳的1s的x射线光电子能谱光谱的反卷积的图7的c)所示,可见碳的1s的峰值分别由相当于c-c(sp2)、c-n、c-o及c=o基的284.8ev、285.9ev、286.6ev及相当于287.9ev的4个峰值形成。在n-cdot/ptpd复合体中,与掺氮碳点的x射线光电子能谱光谱相比(数据未图示),与c-o和c=o之类的碳氧化物相关的峰值大大减少,当制备n-cdot/pt53pd47复合体时,在还原过程中掺氮碳点也一起还原一定程度。
实施例3:n-cdot/ptpd复合体的电化学有效表面积(ecsa)的测定
使用三电极电解电池,通过chi760d电化学工作站(electrochemicalworkstation)(ch仪器公司(instruments,inc.))测定循环伏安法(cyclicvoltammetry)和计时电流法(chronoampemetry)。作为三电极电解电池,将铂线用作对电极,将ag/agcl(饱和nacl)用作基准电极,并将在实施例1中制备的n-cdot/ptpd纳米复合体用作工作电极。n-cdot/ptpd纳米复合体的表面在电化学测定之前,利用nafion(5重量百分比)溶液进行涂敷。作为工作电极利用商品化的pt/c(c-pt/c)催化剂的电极通过以下工序制备:一边使玻璃碳电极(直径为3mm的玻璃碳盘以约0.071cm2的面积嵌入于特氟龙气缸内的结构)进行旋转,一边利用0.3μm和0.05μm的al2o3依次研磨,利用二次离子水洗涤三次。将6mg的c-pt/c催化剂(20%的ptonvulcanxc-72,阿法埃莎(alfaaesar))分散于1ml的水/ipa/nafion离聚物(ionomer)(在79.6∶20(v/v)纳米纯净水和ipa混合物中混合2重量百分比的nagion离聚物溶液(离子功率(ionpower),liquion1100)来制备)溶液,制备油墨。接着,在玻璃碳电极的表面均匀涂敷3μl的催化剂。ptpd/c及多种组成的n-cdot/ptpd纳米复合体催化剂也通过相同的方法以使铂的载量成为35μgcm-2的方式进行制备。
图8为示出上述结果的曲线图,从图8中,实施例的n-cdot/pt84pd16、n-cdot/pt81pd19、n-cdot/pt76p24、n-cdot/pt53pd47及比较例的c-pt/c的电化学表面积(ecsa、electrochemicallyactivesurfacearea)分别计算为89.92、80.37、76.31、53.32及58.03m2/gpt,n-cdot/pt84pd16的电化学表面积比其他多种催化剂明显高。这表示纳米网络结构的球形复合体的表面积宽,电极制备工序中纳米粒子的凝聚少。图8还示出n-cdot/pt84pd16纳米复合体在电化学方面上更敏感,从而可知n-cdot/pt84pd16纳米复合体比其他更有效地接近。这种特性在电子催化剂反应中非常重要。
实施例4:n-cdot/ptpd复合体的电子催化剂性能评价
1)电子催化剂性能的评价
随着期待实施例3中n-cdot/ptpd复合体的电子催化剂性能优秀,评价n-cdot/ptpd复合体的电子催化剂性能,并与ptpd/c及c-pt/c电子催化剂的性能进行比较。
对于甲醇和甲酸氧化反应的cv曲线使用ag/agcl基准电极,在-0.9v和0.50v之间以50mv/s的扫描速度进行测定。对于甲醇和甲酸氧化反应的计时电流法曲线在-0.30和0.20v进行测定。为了cva及计时电流法的测定,在甲醇氧化反应中使用0.1m的koh+1.0m的ch3oh电解质溶液,在甲酸氧化反应中使用0.1m的hclo4+1.0m的hcooh电解质溶液。在测定之前,各个电解质溶液利用高纯度氮气来最少吹洗30分钟来使用,所有测定在常温下实施。图9为示出根据蚀刻时间的n-cdot/ptpd复合体和对于c-pt/c的cv曲线和氧化电流密度的曲线图,图10为比较n-cdot/pt53pd47纳米复合体和在比较例中制备的ptpd/c电子催化剂的cv曲线来示出的曲线图。并且,将通过上述方法测定的电子催化剂特性整理于下列表2中。
表2
图9的a)、b)和图10的a)示出甲醇氧化反应的结果,图10的a)表示n-cdot/pt53pd47纳米复合体的最大电流密度784.95ma/mgpt相比于比较例中制备的ptpd/c的707.01ma/mgpt高出约11%。尤其,如图9的a)和b)及表2所示,可见n-cdot/pt53pd47纳米复合体的初期mor性能与c-pt/c几乎类似,其最大电流密度稍高于c-pt/c。这是n-cdot/pt53pd47纳米复合体内的掺氮碳点的作用,判断为掺氮碳点不仅可极大化单纯地用于电子移动的纳米大小电子催化剂的表面积,而且提高从反应物至电子催化剂的物质移动(masstransport)。进而,纳米网络结构与宽的表面积一同提供电子催化剂的多个活性位置。图9的a)和b)还示出n-cdot/pt84pd16纳米网络结构的球形复合体的最大电流密度相比于c-pt/c或n-cdot/pt53pd47纳米复合体分别高出1.36倍及1.31倍。并且,与以往报告的石墨烯-钒碳氮化物(graphene-vanadiumcarbonitride)/pt杂交物(hybrid)(j.nanoscale2015,7,1301)、pt/rgo-co3o4(rscadvances2014,4,62793)、树枝状(dendritic)ptpdnanogarland/rgo(j.-j.nanoscale2014,6,5708)及pd/pt核壳纳米花(core-shellnanoflowers)/rgo(chemicalcommunications2015,51,10490)等的其他电子催化剂相比,n-cdot/pt84pd16纳米复合体的最大电流密度明显高。
图11为示出对于上述mor电子催化剂的线性扫描伏安图(lsv,linearscanvoltammogram)的曲线图,表示不仅是各个催化剂的最大电流密度,起始电位也不同。对于ag/agcl基准电极而言,c-pt/c和致密的n-cdot/pt53pd47纳米复合体的起始电位分别为-395mv及-476mv,相反,纳米网络结构的n-cdot/pt84pdl6纳米复合体表示-533mv的起始电位,可见对于n-cdot/pt84pd16纳米复合体的甲醇电子氧化的活性度比c-pt/c或n-cdot/pt53pd47纳米复合体优秀。另一方面,钯的量越增加,二元合金中铂的量越减少,致使整体反应速度变慢,因而具有甲醇脱氢化反应被抑制的倾向。这还可从图11的b)中得以确认,n-cdot/ptpd纳米复合体的最大电位随着ptpd二元合金中钯含量减少而逐渐增加。为此,n-cdot/ptpd纳米复合体的最大电流密度也随着增加蚀刻时间来减少钯含量而明显增加。
图9的c,d及图10的b为示出对于甲酸电子氧化反应的催化剂的性能的曲线图。图9的c及图10的b中所示的cv曲线与根据三种途径(triple-pathway)机制的催化剂的cv曲线非常类似。最主要的峰值(峰值iii)相对于ag/agcl基准电极在于0.4~0.7v范围内,这相当于基于甲酸盐阴离子途径的甲酸氧化。氧化反应包括去除氢来形成甲酸盐离子的反应和向co2的后续氧化反应。正向扫描期间的cv曲线分为两个峰值,即,约0.40v左右的峰值i和约0.70v的峰值ii。峰值i相当于从甲酸去除两个氢原子来生成二氧化碳的直接氧化反应(hcoohco2+2h++2e-),峰值ii相当于通过甲酸的脱氢化来生成作为具有催化剂毒性的中间体的一氧化碳之后,通过进一步的氧化来生成二氧化碳的间接氧化反应(hcoohcoads+h2oco2+2h++2e-)。三个峰值中,由于峰值iii的最大电流密度非常高于其他峰值,因而可视为基于n-cdot/ptpd纳米复合体的甲酸的氧化反应通过甲酸盐离子途径发生。图10的b示出在反向扫描期间n-cdot/pt53pd47纳米复合体的最大电流密度相比于ptpd/c纳米粒子高出34%左右。这种n-cdot/pt84pd16纳米复合体的反向电流密度相比于n-cdot/pt53pd47纳米复合体高出约2.05倍,这视为源自n-cdot/pt84pd16纳米复合体的独特结构。如同甲醇的氧化反应,对于甲酸的氧化反应,n-cdot/ptpd纳米复合体的蚀刻时间越经过,最大电流密度越增加,最大电位也正向移动。
2)持续性评价
对于吸附在铂表面的碳性反应中间体的催化剂的耐中毒性由阴极的最大电流密度与阳极的最大电流密度之比(if/ib)表示。这意味着if/ib越高,催化剂更有效地氧化甲醇,基于吸附于表面的种的中毒性越低。如上述表2所示,可见n-cdot/pt84pd16纳米复合体中,if/ib为6.84,这不仅高于c-pt/c或ptpd/c,还比以往报告的其他电子催化剂卓越。
图12的a为表示对于-0.3v(vsag/agcl)的固定电位中测定的甲醇氧化的最大电流密度与时间关系的曲线图。c-pt/c催化剂随着时间的经过其电流密度快速减少,经过3000秒之后,减少为初期电流密度的30%。相反,n-cdot/pt84pd16纳米复合体在3000秒之后,还残留初期电流密度的72%。可确认甲醇氧化反应初期的催化剂的电流密度的快速减少由coads、coohads及choads之类的反应性中间体的生成引起,n-cdot/pt84pd16纳米复合体不仅催化剂活性优秀,而且稳定性也高。该结果与图8的cv的结果一致。
使用0.1mhclo4+1.0mhcooh溶液在0.2v(vsag/agcl),通过计时电流法重新确认一遍这种电子催化剂的活性和电极物质的稳定性。图12的b为示出其结果的曲线图,尤其,可确认到在50秒之前电流密度快速减少。之所以如此是因为抑制co之类的中间体蓄积于电子催化剂的活性区域而使甲酸进一步氧化的现象。这种电流密度的减少在所有电子催化剂中被观测到,但n-cdot/pt84pd16纳米复合体在所有时间上呈现相比于其他多种电子催化剂高的电流密度。
在常温的n2-饱和0.1m的koh溶液中,以50mv/s的扫描速度反复处理电位循环来重新确认一遍n-cdot/ptpd催化剂的稳定性。图13中,a)c-pt/c、b)~e)为根据蚀刻时间的n-cdot/ptpd纳米复合体的初期及30000秒之后的实验结果,b)~e)的蚀刻时间分别为b)0天、c)1天、d)3天、e)5天。在再活化的电子催化剂的分析中,纳米复合体在30000秒之后,保存原来的电化学表面积的84%,与保存约69%的c-pt/c相比,稳定性高出1.2倍左右(参照表3)。
表3
通过在0.1mkoh+1.0mch3oh溶液中测定cv来评价催化剂的性能,并将其结果示于图14中。如同图13,a)c-pt/c、b)~e)示出分别蚀刻0天、1天、3天及5天的n-cdot/ptpd纳米复合体的结果。相对于甲醇氧化反应,其他多种催化剂的最大电流密度大部分减少15%以上,但再活化的n-cdot/pt84pd16纳米复合体在30000秒之后,其最大电流密度也几乎没有变化。
- 还没有人留言评论。精彩留言会获得点赞!