通过抑制MASP‑2依赖性补体活化治疗弥散性血管内凝血的方法与流程
相关申请的交叉引用
本申请主张2009年10月16日提交的临时申请第61/279279号和2010年4月9日提交的临时申请第61/322722号的权益,两者均以引用方式全部并入本文。
关于序列表的声明
与本申请相关联的序列表以文本格式代替纸印本提供,并且在此以引用方式并入本说明书。包含该序列表的文本文件的名称为35668_Seq_Final.txt。该文本文件为109KB;创建于2010年10月15日;并且正随着本说明书的提交经由EFS-Web提交。
技术领域
本发明涉及免疫领域,具体而言,本发明涉及MBL相关的丝氨酸蛋白酶抑制剂在治疗或预防补体介导的凝血障碍中的用途。
背景技术:
补体系统为启动并增强对微生物感染和其它急性损伤的炎症反应提供了早期的作用机制(M.K.Liszewski和J.P.Atkinson,1993,载于Fundamental Immunology,第三版,W.E.Paul编辑,Raven Press,Ltd.,New York)。尽管补体活化提供了重要的针对潜在病原体的第一线防御,但是促进保护性炎症反应的补体活性也可以表现出对宿主的潜在威胁(K.R.Kalli等,Springer Semin.Immunopathol.15:417-431,1994;B.P.Morgan,Eur.J.Clinical Investig.24:219-228,1994)。例如,C3和C5蛋白水解产物募集并活化嗜中性粒细胞。这些活化细胞不加选择地释放破坏性酶,可能造成器官损伤。此外,补体活化可能导致溶胞的补体成分沉积在附近的宿主细胞以及微生物靶上,导致宿主细胞裂解。
已经表明补体系统促成许多急性和慢性疾病状况的发病机制,所述疾病包括:心肌梗塞、中风之后的血管重建、ARDS、再灌注损伤、败血症性休克、热烧伤之后的毛细血管渗漏、心肺分流术后炎症、移植排斥、类风湿性关节炎、多发性硬化、重症肌无力和阿尔茨海默病。在几乎所有的这些疾病中,补体都不是病因,而是发病机制所涉及的若干因素之一。尽管如此,补体活化可能是重要的病理机制,并在许多这类疾病状况的临床控制中表现出有效之处。对各种疾病状况中补体介导的组织损伤的重要性的增加的认识增强了对有效补体抑制药物的需求。还没有药物获准用于特异性靶向并抑制补体活化的人体用途。
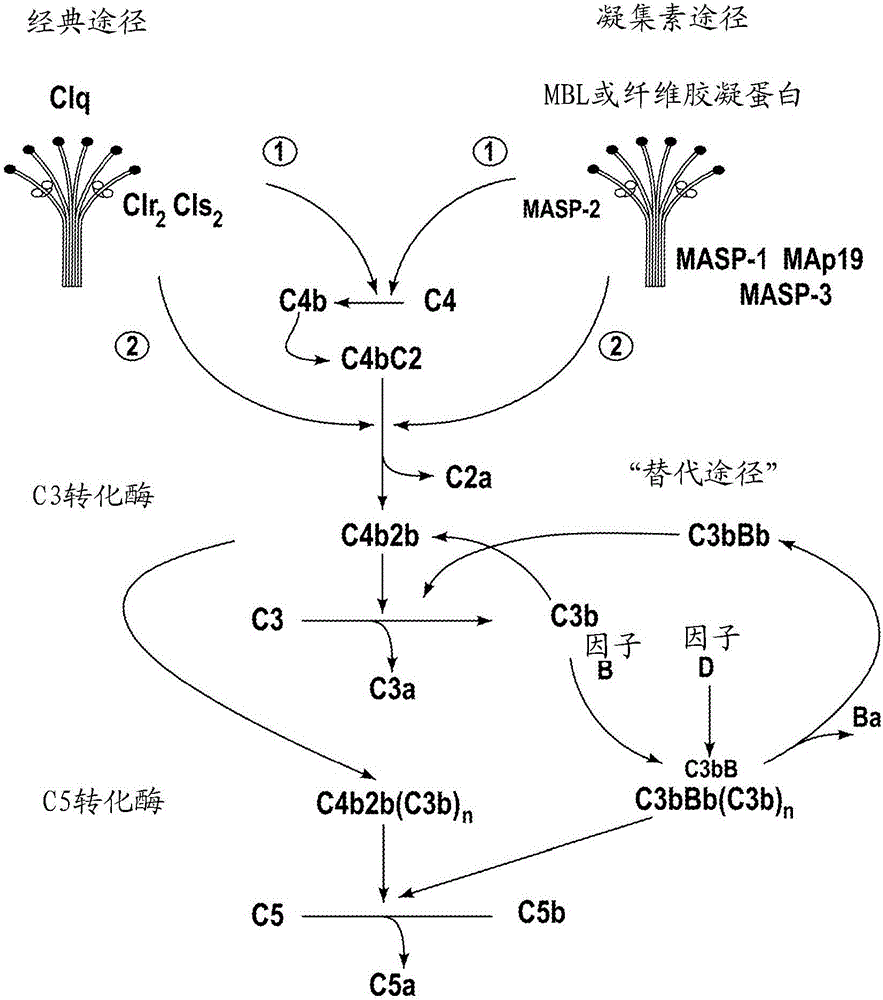
目前,普遍认为补体系统可通过三种截然不同的途径被活化:经典途径、凝集素途径和替代途径。经典途径通常是由抗体与外源颗粒(即抗原)相结合而触发的,因此需要预先暴露于该抗原来产生特异性抗体。因为经典途径的活化与免疫应答的发生有关,所以经典途径是获得性免疫系统的一部分。相反,凝集素途径和替代途径两者不依赖于克隆性免疫(clonal immunity),是先天性免疫系统的一部分。
经典途径活化的第一步是特异性识别分子C1q与结合了抗原的IgG和IgM结合。补体系统的活化导致了丝氨酸蛋白酶酶原的序贯活化。C1q与C1r和C1s丝氨酸蛋白酶酶原结合成称为C1的复合物,当C1q与免疫复合物结合时,C1r的Arg-Ile位点进行自我蛋白酶解,接着是C1r激活C1s,从而获得裂解C4和C2的能力。C4裂解成两个片段,称为C4a和C4b,这使得C4b片段能够与邻近的羟基或氨基形成共价键,随后与活化C2的C2b片段进行非共价相互作用而产生C3转化酶(C4b2b)。C3转化酶(C4b2b)激活C3,导致C5转化酶(C4b2b3b)的产生以及能够引起微生物裂解的膜攻击复合物(C5b-9)的形成。C3和C4的活化形式(C3b和C4b)共价沉积在外源靶表面上,其被多种吞噬细胞上的补体受体所识别。
独立地,补体系统通过凝集素途径活化的第一步也是特异性识别分子的结合,接着是所结合的丝氨酸蛋白酶的活化。然而,凝集素途径中的识别分子是糖结合蛋白(甘露聚糖结合凝集素(MBL)、H-纤维胶凝蛋白(H-ficolin)、M-纤维胶凝蛋白和L-纤维胶凝蛋白),而不是通过C1q来结合免疫复合物(J.Lu等,Biochim.Biophys.Acta1572:387-400,2002;Holmskov等,Annu.Rev.Immunol.21:547-578(2003);Teh等,Immunology 101:225-232(2000))。Ikeda等人首先证明,与C1q类似,MBL在与酵母甘露聚糖包被的红细胞结合时能够以依赖C4的方式使补体系统活化(K.Ikeda等,J.Biol.Chem.262:7451-7454,1987)。MBL是胶原凝集素蛋白家族的成员,是钙依赖性凝集素,与具有定位于吡喃糖环赤道面上的3-羟基和4-羟基的糖类结合。因此MBL的重要配体是D-甘露糖和N-乙酰-D-葡糖胺,而不符合这种空间要求的糖类则对MBL没有可检出的亲和性(Weis,W.I.等,Nature 360:127-134,1992)。MBL和单价糖之间的相互作用是相当微弱的,解离常数通常在2mM的范围内。MBL通过同时与多个单糖残基相互作用来达到对聚糖配体特异性地紧密结合(Lee,R.T.等,Archiv.Biochem.Biophys.299:129-136,1992)。MBL识别通常修饰微生物如细菌、酵母、寄生虫和某些病毒的糖模式。相反,MBL不识别D-半乳糖和唾液酸,即倒数第二位的糖和倒数第一位的糖,它们一般修饰哺乳动物血浆和细胞表面糖蛋白上存在的“成熟”糖缀合物。一般认为这种结合特异性有助于保护免于自身活化。然而,MBL确实以高亲和力结合高甘露糖“前体”聚糖簇,这些簇位于被隔离在哺乳动物细胞内质网和高尔基体内的N-连接的糖蛋白和糖脂上(Maynard,Y.等,J.Biol.Chem.257:3788-3794,1982)。因此,受损细胞是通过MBL结合的凝集素途径活化的潜在目标。
纤维胶凝蛋白(ficolin)具有不同类型的不是MBL的凝集素结构域,称为血纤蛋白原样结构域。纤维胶凝蛋白以不依赖Ca++的方式来结合糖残基。在人体中,已经鉴定出纤维胶凝蛋白的三种类型,即L-纤维胶凝蛋白、M-纤维胶凝蛋白和H-纤维胶凝蛋白。L-纤维胶凝蛋白和H-纤维胶凝蛋白这两种血清纤维胶凝蛋白共同对N-乙酰-D-葡糖胺具有特异性;然而,H-纤维胶凝蛋白还结合N-乙酰-D-半乳糖胺。L-纤维胶凝蛋白、H-纤维胶凝蛋白和MBL的糖特异性的差异意味着不同的凝集素可能是互补的,尽管有重叠,但是可能靶向不同的糖缀合物。这个观点得到了最新报道的支持,即在凝集素途径的已知凝集素中,只有L-纤维胶凝蛋白与脂磷壁酸特异性结合,脂磷壁酸是一种存在于所有革兰氏阳性菌上的细胞壁糖缀合物(Lynch,N.J.等,J.Immunol.172:1198-1202,2004)。胶原凝集素(即MBL)和纤维胶凝蛋白在氨基酸序列上没有显著的相似性。然而,两组蛋白质具有类似的结构域组构,与C1q类似,装配成寡聚结构,这样就使得多位点结合的可能性最大化。MBL的血清浓度在健康人群中是高度可变的,这在遗传上是由MBL基因的启动子和编码区二者的多态性/突变所控制的。作为急性期蛋白,MBL的表达在炎症期间进一步上调。L-纤维胶凝蛋白在血清中存在的浓度与MBL类似。因此,凝集素途径的L-纤维胶凝蛋白分途径在力量上可能与MBL分途径不相上下。MBL和纤维胶凝蛋白也可能作为调理素起作用,这需要这些蛋白质与吞噬细胞受体相互作用(Kuhlman,M.等,J.Exp.Med.169:1733,1989;Matsushita,M.等,J.Biol.Chem.271:2448-54,1996)。然而,吞噬细胞上受体的身份还未得到确定。
人MBL通过其胶原样结构域与独特的C1r/C1s样丝氨酸蛋白酶形成特异性、高亲和性的相互作用,称为MBL相关的丝氨酸蛋白酶(MBL-associated serine proteases,MASP)。迄今为止已经描述了三种MASP。首先,鉴定出单一的酶“MASP”,其特征是负责启动补体级联(即切割C2和C4)的酶(Ji,Y.H.等,J.Immunol.150:571-578,1993)。随后,发现MASP实际上是两种蛋白酶MASP-1和MASP-2的混合物(Thiel,S.等,Nature 386:506-510,1997)。然而,有研究表明仅MBL-MASP-2复合物就足以使补体活化(Vorup-Jensen,T.等,J.Immunol.165:2093-2100,2000)。此外,只有MASP-2快速切割C2和C4(Ambrus,G.等,J.Immunol.170:1374-1382,2003)。因此,MASP-2是负责激活C4和C2以产生C3转化酶C4b2b的蛋白酶。这是不同于C1复合物的显著差异,C1复合物中两种特异性丝氨酸蛋白酶(C1r和C1s)协同作用导致了补体系统的活化。最近,已经分离出第三种新的蛋白酶MASP-3(Dahl,M.R.等,Immunity 15:127-35,2001)。MASP-1和MASP-3是同一基因的可变剪接产物。MASP-1和MASP-3的生物学功能仍有待阐明。
MASP与C1复合物的酶成分C1r和C1s共享相同的结构域组构(Sim,R.B.等,Biochem.Soc.Trans.28:545,2000)。这些结构域包括N末端C1r/C1s/海胆Vegf/骨形成蛋白(CUB)结构域、表皮生长因子样结构域、第二CUB结构域、串联的补体调控蛋白结构域和丝氨酸蛋白酶结构域。与在C1蛋白酶中一样,MASP-2的活化通过丝氨酸蛋白酶结构域附近的Arg-Ile键裂解而产生,这将酶分成二硫键连接的A链和B链,后者由丝氨酸蛋白酶结构域构成。最近,描述了MASP-2的遗传上决定的缺陷(Stengaard-Pedersen,K.等,New Eng.J.Med.349:554-560,2003)。单个核苷酸的突变导致CUB 1结构域的Asp-Gly交换,并使得MASP-2不能与MBL结合。
MBL还与称为19kDa MBL相关蛋白(MAp19)(Stover,C.M.,J.Immunol.162:3481-90,1999)或者小MBL相关蛋白(sMAP)(Takahashi,M.等,Int.Immunol.11:859-863,1999)的非酶蛋白缔合。MAp19通过MASP 2基因产物的可变剪接而产生,包括MASP-2的头两个结构域,其后接着是4个独特氨基酸的额外序列。MASP 1和MASP 2基因分别位于3号染色体和1号染色体上(Schwaeble,W.等,Immunobiology 205:455-466,2002)。
若干证据表明存在不同的MBL-MASP复合物,且血清中的总MASP的大部分不与MBL结合(Thiel,S.等,J.Immunol.165:878-887,2000)。H-纤维胶凝蛋白和L-纤维胶凝蛋白都与MASP结合,并且激活凝集素补体途径,同MBL一样(Dahl,M.R.等,Immunity15:127-35,2001;Matsushita,M.等,J.Immunol.168:3502-3506,2002)。凝集素途径和经典途径都形成共同的C3转化酶(C4b2b),这两条途径在这一步会合。
普遍认为凝集素途径在宿主抵抗感染的防御中具有重要作用。MBL参与宿主防御的强有力证据来自于对功能性MBL血清水平降低的患者的分析(Kilpatrick,D.C.,Biochim.Biophys.Acta 1572:401-413,2002)。这些患者表现出复发性细菌和真菌感染的易感性。这些症状通常在生命早期在易损表观窗期间是明显的,因为从母体获得的抗体效价降低,但处于全部抗体应答发展之前。这种症状经常是由于MBL胶原部分的数个位点突变引起的,其妨碍了MBL寡聚体的正确形成。然而,由于MBL可以作为不依赖于补体的调理素起作用,所以还不知道对感染的易感性增加在多大程度上是由于受损的补体活化所致。
尽管大量证据表明在非感染性人类疾病的发病机制中,经典补体途径和替代补体途径两者都存在,但是对凝集素途径作用的评价才刚刚开始。最新研究提供的证据表明,凝集素途径的活化可能是造成缺血/再灌注损伤中补体活化和相关炎症的原因。Collard等人(2000)报告受到氧化应激的培养的内皮细胞结合MBL,且在暴露于人血清时显示出C3沉积(Collard,C.D.等,Am.J.Pathol.156:1549-1556,2000)。此外,用封闭性抗MBL单克隆抗体处理人血清抑制了MBL结合和补体活化。将这些发现扩展到心肌缺血-再灌注大鼠模型上,其中比起用对照抗体处理的大鼠,用针对大鼠MBL的封闭性抗体处理的大鼠在冠状动脉闭塞时显示心肌损伤显著较轻(Jordan,J.E.等,Circulation 104:1413-1418,2001)。尚不清楚氧化应激后MBL与血管内皮结合的分子机制;最近的研究表明,氧化应激后凝集素途径的活化可能是由MBL与血管内皮细胞角蛋白结合而介导的,而不是与糖缀合物结合(Collard,C.D.等,Am.J.Pathol.159:1045-1054,2001)。其它研究已显示出缺血/再灌注损伤发病机制中的经典途径和替代途径以及凝集素途径在这种疾病中的作用仍然存在争议(Riedermann,N.C.等,Am.J.Pathol.162:363-367,2003)。
与经典途径和凝集素途径相反,没有发现替代途径中完成识别功能的起始因子(initiator),而在其它两种途径中是C1q和凝集素来完成识别功能的。目前普遍接受的是,替代途径是由外来表面或其它异常表面(细菌、酵母、病毒感染细胞或者受损组织)自发触发的。有四种血浆蛋白直接参与了替代途径:C3、B因子、D因子和备解素。对于替代途径,需要由天然C3蛋白水解形成的C3b发挥作用。由于替代途径C3转化酶(C3bBb)含有C3b作为必需亚基,关于通过替代途径的第一个C3b来源的问题代表了令人困扰的问题,且促使了相当多的研究工作。
C3属于含有极少的被称为硫酯键的翻译后修饰的蛋白质家族(还有C4和α-2巨球蛋白)。硫酯基团由谷氨酰胺的末端羰基与三个氨基酸距离的半胱氨酸的巯基相结合所形成。该键不稳定,且谷氨酰胺的亲电子羰基可与其它分子通过羟基或氨基形成共价键。当被隔离在完整C3的疏水口袋内部时,硫酯键是相当稳定的。然而,C3被蛋白酶切割成C3a和C3b,导致C3b上高反应性的硫酯键暴露出来,通过该机制C3b与靶共价结合。除了有大量文件证明C3b与补体靶共价结合的作用外,还有研究认为C3硫酯具有触发替代途径的关键作用。根据普遍接受的“慢转理论(tick-over theory)”,替代途径是由液相转化酶iC3Bb的形成所启动的,iC3Bb是由C3与水解硫酯(iC3;C3(H2O))和B因子形成的(Lachmann,P.J.等,Springer Semin.Immunopathol.7:143-162,1984)。C3b样iC3由天然C3经蛋白质中内部硫酯的缓慢自发水解而产生(Pangburn,M.K.等,J.Exp.Med.154:856-867,1981)。通过iC3Bb转化酶的活性,C3b分子沉积在靶表面,从而启动替代途径。
对替代途径活化的起始因子的了解甚少。认为激活因子包括酵母细胞壁(酵母聚糖)、许多纯的多糖、兔红细胞、某些免疫球蛋白、病毒、真菌、细菌、动物肿瘤细胞、寄生虫和受损细胞。这些激活因子所共有的唯一特征是糖类的存在,但是糖类结构的复杂性和多样性使得难以确定共享的分子决定子,这是公认的。
对于凝集素/经典途径C3转化酶(C4b2b),替代途径也可以提供强大的放大环路(amplification loop),因为所产生的任何C3b都能与B因子一起参与形成额外的替代途径C3转化酶(C3bBb)。替代途径C3转化酶通过备解素的结合而稳定。备解素延长了替代途径C3转化酶的半衰期6-10倍。向C3转化酶中添加C3b导致替代途径C5转化酶的形成。
一直以来认为所有三种途径(即经典、凝集素和替代途径)会合于C5,它被裂解形成具有多种促炎作用的产物。会合后的途径被称为末端补体途径。C5a是最有效的过敏毒素,引起平滑肌和血管紧张度以及血管通透性的改变。它也是嗜中性粒细胞和单核细胞两者的强有力的趋化因子和激活因子。C5a介导的细胞活化能够通过诱导释放多种另外的炎症介质来显著放大炎症反应,另外的炎症介质包括细胞因子、水解酶、花生四烯酸代谢物和活性氧类别。C5裂解导致了C5b-9的形成,它也被称为膜攻击复合物(MAC)。目前强有力的证据表明,亚裂解的MAC沉积除了起裂解成孔复合物的作用外,还可能还在炎症中发挥重要作用。
技术实现要素:
提供本概述,从而以简化形式引入概念的选择,其在以下发明详述中进一步描述。本概述既不欲鉴定请求保护的主题的关键特征,也不欲用于协助确定请求保护的主题的范围。
一方面,本发明提供抑制有生命的受试者中MASP-2依赖性补体活化的不利作用的方法。该方法包括将有效抑制MASP-2依赖性补体活化的量的MASP-2抑制剂给予有需要的受试者的步骤。在本上下文中,短语“MASP-2依赖性补体活化”是指通过凝集素依赖性MASP-2系统而发生的替代途径补体活化。在本发明的另一方面,MASP-2抑制剂通过凝集素依赖性MASP-2系统抑制补体活化,但基本上不抑制通过经典或C1q依赖性系统的补体活化,从而C1q依赖性系统仍然具有功能。
在本发明这些方面的一些实施方案中,MASP-2抑制剂是抗MASP-2抗体或其片段。在其它实施方案中,抗MASP-2抗体具有降低的效应子功能。在一些实施方案中,MASP-2抑制剂是MASP-2抑制肽或非肽MASP-2抑制剂。
另一方面,本发明提供用于抑制MASP-2依赖性补体活化的不利作用的组合物,该组合物包含治疗有效量的MASP-2抑制剂和药学上可接受的载体。本发明还提供制备用于抑制有需要的有生命的受试者中MASP-2依赖性补体活化的不利作用的药物的方法,其包括药物载体中的治疗有效量的MASP-2抑制剂。本发明还提供制备用于抑制MASP-2依赖性补体活化以治疗下文所述病症、疾病和障碍中每一种的药物的方法。
本发明的方法、组合物和药物用于在患有如本文将进一步描述的急性或慢性病理病症或损伤的哺乳动物受试者、包括人中体内抑制MASP-2依赖性补体活化的不利作用。这些疾病和损伤包括但不限于与自身免疫性疾病和/或炎性疾病相关的MASP-2介导的补体活化。
在本发明的另一个方面,提供用于抑制受试者中MASP-2依赖性补体活化的方法,所述受试者患有凝血障碍如补体介导的凝血障碍或凝血病、或处于发生凝血障碍如补体介导的凝血障碍或凝血病的风险中,所述方法通过向该受试者给予药物载体中的治疗有效量的MASP-2抑制剂来实施。接受根据本发明的治疗的病症包括(借助于非限制性实例)弥散性血管内凝血("DIC"),也被称作消耗性凝血病。
在本发明的一个方面,提供用于抑制受试者中MASP-2依赖性补体活化的方法,所述受试者患有凝血障碍或处于发生凝血障碍的风险中,所述方法包括:给予该受试者有效抑制MASP-2依赖性补体活化的量的MASP-2抑制剂。
在本发明的另一个方面,提供用于抑制受试者中MASP-2依赖性补体活化的方法,所述受试者患有凝血障碍或处于发生凝血障碍的风险中,所述方法包括:给予该受试者有效选择性抑制MASP-2依赖性补体活化而基本上不抑制C1q依赖性补体活化的量的MASP-2抑制剂。
在本发明的另一个方面,提供制备用于抑制患有补体介导的凝血障碍的有生命的受试者中MASP-2依赖性补体活化的作用的药物的方法,所述方法包括:将治疗有效量的MASP-2抑制剂组合在药物载体中。
在本发明的另一个方面,提供用于在有需要的受试者中治疗弥散性血管内凝血、预防弥散性血管内凝血或降低弥散性血管内凝血的严重程度的方法,所述方法包括:给予该受试者包含有效抑制MASP-2依赖性补体活化的量的MASP-2抑制剂的组合物。
附图说明
通过参考下面的详细描述连同附图,本发明前述的方面以及许多附带的优点将更容易领会,同样也更好理解,其中:
图1是图解替代补体途径的补体活化需要凝集素途径依赖的MASP-2活化这一新发现的流程图;
图2是图解人MASP-2基因组结构的图;
图3A是图解人MASP-2蛋白的结构域结构的示意图;
图3B是图解人MAp19蛋白的结构域结构的示意图;
图4是图解鼠MASP-2敲除策略的图;
图5是图解人MASP-2微基因构建体的图;
图6A提供了证明MASP-2缺乏导致凝集素途径介导的C4活化丧失的结果,如通过甘露聚糖上C4b沉积缺乏所测定的;
图6B提供了证明MASP-2缺乏导致凝集素途径介导的C4活化丧失的结果,如通过酵母聚糖上C4b沉积缺乏所测定的;
图6C提供了证明获自MASP-2+/-、MASP-2-/-和野生型品系的血清样品的相对C4活化水平的结果,如通过甘露聚糖和酵母聚糖上的C4b沉积所测定的;
图7A提供了证明MASP-2缺乏导致凝集素途径介导的以及替代途径介导的C3活化丧失的结果,如通过甘露聚糖上C3b沉积缺乏所测定的;
图7B提供了证明MASP-2缺乏导致凝集素途径介导的以及替代途径介导的C3活化丧失的结果,如通过酵母聚糖上C3b沉积缺乏所测定的;
图8提供了证明加入鼠重组MASP-2到MASP-2-/-血清样品中以蛋白浓度依赖的方式恢复了凝集素途径介导的C4活化的结果,如通过甘露聚糖上的C4b沉积所测定的;
图9提供了证明MASP-2-/-品系中经典途径有功能的结果;
图10提供了证明MASP-2依赖的补体活化系统在腹主动脉瘤修复之后在缺血/再灌注阶段中活化的结果;
图11A提供了证明抗MASP-2Fab2抗体#11抑制C3转化酶形成的结果,如实施例24所述;
图11B提供了证明抗MASP-2Fab2抗体#11与天然大鼠MASP-2结合的结果,如实施例24所述;
图11C提供了证明抗MASP-2Fab2抗体#41抑制C4裂解的结果,如实施例24所述;
图12提供了证明经测试抑制C3转化酶形成的所有抗MASP-2Fab2抗体还被发现抑制C4裂解的结果,如实施例24所述;
图13是表示从用于抗MASP-2封闭性Fab2抗体表位作图的大鼠MASP-2衍生的重组多肽的示意图,如实施例25所述;
图14提供了证明抗MASP-2Fab2#40和#60与大鼠MASP-2多肽结合的结果,如实施例25所述;
图15提供了证明肾缺血/再灌注损伤模型中再灌注后24小时和48小时野生型(+/+)和MASP-2(-/-)小鼠的血尿素氮清除率的结果,如实施例26所述;
图16A提供了证明冠状动脉闭塞和再灌注模型中损伤后野生型(+/+)的梗塞大小及MASP-2(-/-)小鼠中梗塞大小减小的结果,如实施例27所述;
图16B提供了显示冠状动脉闭塞和再灌注模型中各受试动物的分布的结果,如实施例27所述;
图17A提供了显示从野生型(+/+)和MASP-2(-/-)小鼠中分离的RPE-脉络膜复合体中的基线VEGF蛋白水平的结果,如实施例28所述;
图17B提供了显示黄斑变性模型中激光诱发的损伤之后第三天野生型(+/+)和MASP-2(-/-)小鼠的RPE-脉络膜复合体中VEGF蛋白水平的结果,如实施例28所述;
图18提供了显示野生型(+/+)和MASP-2(-/-)小鼠中激光诱发损伤后第七天平均脉络膜新血管形成(CNV)体积的结果,如实施例28所述;
图19提供了显示野生型(+/+)和MASP-2(-/-)小鼠在Col2mAb诱发类风湿性关节炎后随时间变化的平均临床关节炎评分的结果,如实施例29所述;
图20A是显示sMAP(Map 19)基因定向破坏的示意图,如实施例30所述;
图20B提供了从得自雄性sMAP(-/-)嵌合小鼠与雌性C57Bl/6小鼠交配的子代分离的基因组DNA的DNA印迹分析,如实施例30所述;
图20C提供了野生型(+/+)和sMAP(-/-)小鼠的PCR基因型分型分析,如实施例30所述;
图21A提供了sMAP(-/-)小鼠中sMAP和MASP-2mRNA的RNA印迹分析,如实施例30所述;
图21B提供了野生型(+/+)和sMAP(-/-)小鼠中编码MASP-2H链、MASP-2L链和sMAP的cDNA的定量RT-PCR分析,如实施例30所述;
图22A提供了sMAP(-/-)即MAp 19(-/-)的免疫印迹,证实小鼠血清中缺乏MASP-2和sMAP,如实施例30所述;
图22B提供了证明在MBL-MASP-sMAP复合物中检出MASP-2和sMAP的结果,如实施例30所述;
图23A提供了显示野生型(+/+)和sMAP(-/-)小鼠血清中C4沉积在甘露聚糖包被的孔中的结果,如实施例30所述;
图23B提供了显示野生型(+/+)和sMAP(-/-)小鼠血清中C3沉积在甘露聚糖包被的孔中的结果,如实施例30所述;
图24A提供了显示sMAP(-/-)血清中的MBL-MASP-sMAP复合物的重构的结果,如实施例30所述;
图24B-D提供了显示rsMAP和MASP-2i竞争性结合MBL的结果,如实施例30所述;
图25A-B提供了显示通过加入rMASP-2而不是rsMAP使C4沉积活性恢复的结果,如实施例30所述;
图26A-B提供了显示通过加入rsMAP来降低C4沉积活性的结果,如实施例30所述;
图27A-C提供了显示MASP-2是造成C3的C4旁路活化的原因的结果,如实施例31所述;
图28A和图28B提供了在正常大鼠血清中给予MASP-2Fab2抗体之后C4b沉积的抑制(图28A)和凝血酶活化的抑制的剂量响应曲线,如实施例32中所述;
图29A和图29B提供了在弥散性血管内凝血的局部Schwartzman反应模型中,与未处理的野生型小鼠和其中补体途径被放血剂眼镜蛇毒因子(cobra venom factor,CVF)和终末途径抑制剂(C5aR拮抗剂)抑制的野生型小鼠中的血小板凝集(图29A)相比,MASP-2(-/-)小鼠中测定的血小板凝集(表示为凝集面积)(图29B),如实施例33中所述;
图30A-30C图示在特异于经典或凝集素途径激活路线的试验中,C4-/-血浆中的C3周转的研究结果;
图31A将平均危险区(area-at-risk,AAR)和梗塞体积(INF)图示为在经历冠状动脉左前降支闭塞和再灌注之后WT(+/+)和MASP-2(-/-)小鼠中的总心肌体积的百分比,如实施例34中所述;
图31B将梗塞体积(INF)与平均危险区(AAR)之间的关联图示为在经历动脉闭塞和再灌注之后WT(+/+)和MASP-2(-/-)小鼠中的左心室心肌体积的百分比,如实施例34中所述;
图31C图示根据Langendorff分离-灌注小鼠心脏模型制备的WT(+/+)和MASP-2(-/-)小鼠的缓冲液灌注心脏中的梗塞体积(INF),其中全心缺血和再灌注在无血清的情况下进行,如实施例34中所述;
图31D图示根据Langendorff分离-灌注小鼠心脏模型制备的WT(+/+)和MASP-2(-/-)小鼠的缓冲液灌注心脏中的梗塞体积(INF)与风险区带之间的关联,如实施例34中所述;
图32图示在WT(+/+)供体肾的WT(+/+)(B6)或MASP-2(-/-)移植受体小鼠中测定的血尿素氮(BUN)水平,如实施例35中所述;
图33将WT(+/+)和MASP-2(-/-)小鼠的存活者百分比图示为盲肠结扎和穿刺(CLP)模型中微生物感染后的天数的函数,如实施例36中所述;
图34图示盲肠结扎和穿刺(CLP)模型中微生物感染后在WT(+/+)和MASP-2(-/-)中测定的细菌数,如实施例36中所述;
图35为Kaplan-Mayer曲线,该曲线图示在用绿脓假单胞菌的鼻内给药激发后6天WT(+/+)、MASP-2(-/-)和C3(-/-)小鼠的存活百分比,如实施例37中所述;
图36图示在WT小鼠内小鼠抗MASP-2单克隆抗体的0.3mg/kg或1.0mg/kg皮下给药之后,在不同时间点取样的样品中C4b沉积的水平,测定为对照的%,如实施例38中所述;
图37图示在WT小鼠内小鼠抗MASP-2单克隆抗体的0.6mg/kg腹膜内给药之后,在不同时间点取样的样品中C4b沉积的水平,测定为对照的%,如实施例38中所述;
图38图示在用0.3mg/kg或1.0mg/kg小鼠抗MASP-2单克隆抗体的单次腹膜内注射预处理的WT(+/+)小鼠中激光诱发损伤后第7天的平均脉络膜新生血管形成(CNV)体积,如实施例39中所述;
图39A图示在用5×108/100μl脑膜炎奈瑟氏菌(N.meningitidis)感染后MASP-2(-/-)和WT(+/+)小鼠的存活百分比,如实施例40中所述;
图39B图示采自用5×108cfu/100μl脑膜炎奈瑟氏菌感染的MASP-2KO(-/-)和WT(+/+)小鼠的血样中在不同时间点重新获得的脑膜炎奈瑟氏菌的log cfu/ml,如实施例40中所述;
图40A图示在用2×108cfu/100μl脑膜炎奈瑟氏菌感染后MASP-2KO(-/-)和WT(+/+)小鼠的存活百分比,如实施例40中所述;
图40B图示采自用2×108cfu/100μl脑膜炎奈瑟氏菌感染的WT(+/+)小鼠的血样中在不同时间点重新获得的脑膜炎奈瑟氏菌的log cfu/ml,如实施例40中所述;以及
图40C图示采自用2×108cfu/100μl脑膜炎奈瑟氏菌感染的MASP-2(-/-)小鼠的血样中在不同时间点重新获得的脑膜炎奈瑟氏菌的log cfu/ml,如实施例40中所述。
序列表描述
SEQ ID NO:1人MAp 19cDNA
SEQ ID NO:2人MAp 19蛋白(带前导序列)
SEQ ID NO:3人MAp 19蛋白(成熟)
SEQ ID NO:4人MASP-2cDNA
SEQ ID NO:5人MASP-2蛋白(带前导序列)
SEQ ID NO:6人MASP-2蛋白(成熟)
SEQ ID NO:7人MASP-2gDNA(外显子1-6)
抗原:(关于MASP-2成熟蛋白)
SEQ ID NO:8CUBI序列(氨基酸1-121)
SEQ ID NO:9CUBEGF序列(氨基酸1-166)
SEQ ID NO:10CUBEGFCUBII(氨基酸1-293)
SEQ ID NO:11EGF区(氨基酸122-166)
SEQ ID NO:12丝氨酸蛋白酶结构域(氨基酸429-671)
SEQ ID NO:13无活性的丝氨酸蛋白酶结构域(氨基酸610-625,具有Ser618至Ala的突变)
SEQ ID NO:14TPLGPKWPEPVFGRL(CUB1肽)
SEQ ID NO:15TAPPGYRLRLYFTHFDLELSHLCEYDFVKLSS GAKVLATLCGQ(CUBI肽)
SEQ ID NO:16TFRSDYSN(MBL结合区核心)
SEQ ID NO:17FYSLGSSLDITFRSDYSNEKPFTGF(MBL结合区)
SEQ ID NO:18IDECQVAPG(EGF肽)
SEQ ID NO:19ANMLCAGLESGGKDSCRGDSGGALV(丝氨酸蛋白酶结合核心)详述
肽抑制剂:
SEQ ID NO:20MBL全长cDNA
SEQ ID NO:21MBL全长蛋白
SEQ ID NO:22OGK-X-GP(共有结合序列)
SEQ ID NO:23OGKLG
SEQ ID NO:24GLR GLQ GPO GKL GPO G
SEQ ID NO:25GPO GPO GLR GLQ GPO GKL GPO GPO GPO
SEQ ID NO:26GKDGRDGTKGEKGEPGQGLRGLQGPOGKL GPOG
SEQ ID NO:27GAOGSOGEKGAOGPQGPOGPOGKMGPKGEO GDO(人h-纤维胶凝蛋白)
SEQ ID NO:28GCOGLOGAOGDKGEAGTNGKRGERGPOGPO GKAGPOGPNGAOGEO(人纤维胶凝蛋白p35)
SEQ ID NO:29LQRALEILPNRVTIKANRPFLVFI(C4裂解位点)
表达抑制剂:
SEQ ID NO:30CUBI-EGF结构域的cDNA(SEQ ID NO:4的核苷酸22-680)
SEQ ID NO:31
5'CGGGCACACCATGAGGCTGCTGACCCTCC TGGGC 3'SEQ ID NO:4的核苷酸12-45,包括MASP-2翻译起始位点(有义链)
SEQ ID NO:32
5'GACATTACCTTCCGCTCCGACTCCAACGAG AAG 3',编码含有MASP-2MBL结合位点区域的SEQ ID NO:4的核苷酸361-396(有义链)
SEQID NO:33
5'AGCAGCCCTGAATACCCACGGCCGTATCCC AAA 3',编码含有CUBII结构域区域的SEQ ID NO:4的核苷酸610-642
克隆引物:
SEQ ID NO:34CGGGATCCATGAGGCTGCTGACCCTC(用于CUB的5'PCR)
SEQ ID NO:35GGAATTCCTAGGCTGCATA(用于CUB的3'PCR)
SEQ ID NO:36GGAATTCCTACAGGGCGCT(用于CUBIEGF的3'PCR)
SEQ ID NO:37GGAATTCCTAGTAGTGGAT(用于CUBIEGFCUBII的3'PCR)
SEQ ID NO:38-47是人源化抗体的克隆引物
SEQ ID NO:48是9个氨基酸的肽键
表达载体:
SEQ ID NO:49是MASP-2小基因插入序列
SEQ ID NO:50是鼠MASP-2cDNA
SEQ ID NO:51是鼠MASP-2蛋白(w/前导序列)
SEQ ID NO:52是成熟鼠MASP-2蛋白
SEQ ID NO:53是大鼠MASP-2cDNA
SEQ ID NO:54是大鼠MASP-2蛋白(w/前导序列)
SEQ ID NO:55是成熟大鼠MASP-2蛋白
SEQ ID NO:56-59是用于人MASP-2的定点诱变的寡核苷酸,所述人MASP-2用来产生人MASP-2A
SEQ ID NO:60-63是用于鼠MASP-2的定点诱变的寡核苷酸,所述鼠MASP-2用来产生鼠MASP-2A
SEQ ID NO:64-65是用于大鼠MASP-2的定点诱变的寡核苷酸,所述大鼠MASP-2用来产生大鼠MASP-2A。
具体实施方式
本发明基于本发明的发明人预料不到的发现,即需要MASP-2来启动替代补体途径的活化。通过使用敲除小鼠模型MASP-2-/-,本发明的发明人已显示,通过凝集素介导的MASP-2途径抑制替代补体途径活化而同时保持经典途径的完整是可能的,从而确定了凝集素依赖性MASP-2活化是缺少经典途径的替代补体活化所需要的。本发明也描述了MASP-2作为治疗靶的用途,用于抑制与凝集素介导的替代补体途径活化有关的细胞损伤而同时保持免疫系统经典(C1q依赖性的)途径成分的完整。
I.定义
除非本文明确规定,否则本文使用的所有术语都具有如本发明领域的普通技术人员所理解的相同含义。为了明确用于本说明书和所附权利要求书中描述本发明的有关术语,提供下列定义。
本文所用术语“MASP-2依赖性补体活化”是指经凝集素依赖性MASP-2活化而发生的替代途径补体活化。
本文所用术语“替代途径”是指例如由酵母聚糖触发的补体活化,所述酵母聚糖来自真菌和酵母细胞壁、革兰氏阴性外膜的脂多糖(LPS)和兔红细胞以及来自多种纯的多糖、兔红细胞、病毒、细菌、动物肿瘤细胞、寄生虫和受损细胞,在传统上一直认为是由来自补体因子C3自发水解产生的C3b而引起的。
本文所用术语“凝集素途径”是指通过血清和非血清糖结合蛋白(包括甘露聚糖结合凝集素(MBL)和纤维胶凝蛋白)的特异性结合而发生的补体活化。
本文所用术语“经典途径”是指由抗体与外源颗粒结合而触发的并且需要结合识别分子C1q的补体活化。
本文所用术语“MASP-2抑制剂”是指与MASP-2结合或直接与MASP-2相互作用并有效抑制MASP-2依赖性补体活化的任何试剂,包括抗MASP-2抗体及其MASP-2结合片段、天然和合成肽、小分子、可溶性MASP-2受体、表达抑制剂和分离的天然抑制剂,还包括在凝集素途径中为结合其它识别分子(例如MBL、H-纤维胶凝蛋白、M-纤维胶凝蛋白或L-纤维胶凝蛋白)而与MASP-2竞争的肽,但是不包括与这些其它的识别分子结合的抗体。用于本发明方法的MASP-2抑制剂可降低MASP-2依赖性补体活化大于20%,例如大于50%,例如大于90%。在一个实施方案中,MASP-2抑制剂降低MASP-2依赖性补体活化大于90%(即导致仅仅10%或更低的MASP-2补体活化)。
本文所用术语“抗体”包括得自产生抗体的任何哺乳动物(如小鼠、大鼠、兔和包括人在内的灵长类动物)并与MASP-2多肽或其部分特异性结合的抗体及其抗体片段。示例性的抗体包括多克隆抗体、单克隆抗体和重组抗体;多特异性抗体(如双特异性抗体);人源化抗体;鼠抗体;嵌合的小鼠-人、小鼠-灵长类、灵长类-人单克隆抗体;和抗独特型抗体,且可以是任何完整的分子或其片段。
本文所用术语“抗体片段”是指得自或涉及全长抗MASP-2抗体的一部分,一般包括抗原结合区或其可变区。抗体片段的说明性实例包括Fab、Fab'、F(ab)2、F(ab')2和Fv片段、scFv片段、双抗体、线性抗体、单链抗体分子和由抗体片段形成的多特异性抗体。
本文所用的“单链Fv”或“scFv”抗体片段包括抗体的VH和VL结构域,其中这些结构域存在于单一多肽链上。Fv多肽一般还包括VH与VL结构域之间的多肽接头,这使得scFv能够形成所需的抗原结合结构。
本文所用术语“嵌合抗体”是含有得自非人物种(如啮齿动物)抗体的可变结构域和互补决定区的重组蛋白,而抗体分子的其余部分来源于人抗体。
本文所用术语“人源化抗体”是包含符合源自非人免疫球蛋白的特异性互补决定区的最小序列的嵌合抗体,该特异性互补决定区被植入人抗体构架中。人源化抗体通常是其中只有抗体互补决定区是非人来源的重组蛋白。
本文所用术语“甘露聚糖结合凝集素”(“MBL”)等同于甘露聚糖结合蛋白(“MBP”)。
本文所用的“膜攻击复合物”(“MAC”)是指插入并破坏膜的5种末端补体成分(C5-C9)的复合物。亦称C5b-9。
本文所用的“受试者”包括所有哺乳动物,包括但不限于人、非人灵长类动物、狗、猫、马、绵羊、山羊、牛、兔、猪和啮齿动物。
本文所用的氨基酸残基的缩写如下:丙氨酸(Ala;A)、天冬酰胺(Asn;N)、天冬氨酸(Asp;D)、精氨酸(Arg;R)、半胱氨酸(Cys;C)、谷氨酸(Glu;E)、谷胺酰胺(Gln;Q)、甘氨酸(Gly;G)、组氨酸(His;H)、异亮氨酸(Ile;I)、亮氨酸(Leu;L)、赖氨酸(Lys;K)、甲硫氨酸(Met;M)、苯丙氨酸(Phe;F)、脯氨酸(Pro;P)、丝氨酸(Ser;S)、苏氨酸(Thr;T)、色氨酸(Trp;W)、酪氨酸(Tyr;Y)和丙氨酸(Val;V)。
从最广意义上看,天然存在的氨基酸可根据各个氨基酸侧链的化学特性来分组。“疏水”氨基酸是指Ile、Leu、Met、Phe、Trp、Tyr、Val、Ala、Cys或Pro。“亲水”氨基酸是指Gly、Asn、Gln、Ser、Thr、Asp、Glu、Lys、Arg或His。氨基酸的这种组别可进一步细分如下。“不带电荷的亲水”氨基酸是指Ser、Thr、Asn或者Gln。“酸性”氨基酸是指Glu或Asp。“碱性”氨基酸是指Lys、Arg或者His。
本文所用术语“保守的氨基酸取代”通过下面每组中氨基酸之间的取代来说明:(1)甘氨酸、丙氨酸、缬氨酸、亮氨酸和异亮氨酸;(2)苯丙氨酸、酪氨酸和色氨酸;(3)丝氨酸和苏氨酸;(4)天冬氨酸和谷氨酸;(5)谷胺酰胺和天冬酰胺;(6)赖氨酸、精氨酸和组氨酸。
本文所用术语“寡核苷酸”是指核糖核酸(RNA)或脱氧核糖核酸(DNA)或其模拟物的寡聚体或多聚体。该术语还涵盖由天然存在的核苷酸、糖和核苷间(骨架)共价键以及具有非天然存在的修饰的寡核苷酸所组成的寡核苷酸碱基。
II.替代途径:新见解
补体的替代途径首先是由Louis Pillemer和他的同事在1950年代初期根据由酵母细胞壁制备的酵母聚糖用来激活补体的研究提出的(Pillemer,L.等,J.Exp.Med.103:1-13,1956;Lepow,I.H.,J.Immunol.125:471-478,1980)。从那时起直到现在,酵母聚糖被认为是人和啮齿动物血清中替代途径的特异性激活因子的标准实例(Lachmann,P.J.等,Springer Semin.Immunopathol.7:143-162,1984;Van Dijk,H.等,J.Immunol.Methods 85:233-243,1985;Pangburn,M.K.,Methods in Enzymol.162:639-653,1988)。用于替代途径活化的简便而广泛使用的测定法是将血清与包被在塑料孔中的酵母聚糖一起温育,以测定温育后固相上C3b沉积的量。正如所料,与正常小鼠血清温育之后,在酵母聚糖包被孔中有大量C3b沉积(图7B)。然而,与正常血清相比,得自纯合MASP-2缺陷型小鼠的血清与酵母聚糖包被孔一起温育导致C3b沉积大为减少。此外,在该实验中使用MASP 2基因缺陷型杂合小鼠的血清产生的C3b沉积水平,介于纯合MASP-2缺陷型小鼠血清与正常小鼠血清所得到的C3b沉积水平的中间。使用已知激活替代途径的另一种多糖甘露聚糖包被的孔也获得了类似结果(图7A)。由于除了MASP 2基因之外,正常小鼠和MASP-2缺陷型小鼠共享相同的遗传背景,因此这些预料不到的结果表明,MASP-2在替代途径活化方面起着重要作用。
这些结果提供了强有力的证据,证明替代途径并非象基本上所有现有的有关补体的医学教科书和最新的综述文章中所描述的那样是不受限制的独立的补体活化途径。现行普遍持有的科学观点是,替代途径是在某些微粒靶(微生物、酵母聚糖、兔红细胞)的表面上被自发的“慢转”C3活化的扩大所激活的。然而,在MASP-2敲除小鼠血清中,通过两种众所周知的替代途径“激活因子”都没有产生显著的替代途径活化,这使得无法通过“慢转理论”来解释补体活化的重要生理机制。
由于已知MASP-2蛋白酶具有明确的特异性酶作用,负责凝集素补体级联的启动,因此这些结果提示由酵母聚糖和甘露聚糖导致的凝集素途径的活化是随后替代途径活化的关键性的第一步。C4b是由凝集素途径产生而不是由替代途径产生的活化产物。与这个观点相一致,正常小鼠血清与酵母聚糖或甘露聚糖包被的孔进行温育时导致了C4b沉积在孔上,当包被孔与得自MASP-2缺陷型小鼠的血清温育时,这种C4b沉积大大减少(图6A、图6B和图6C)。
替代途径除了作为补体活化的独立途径这一被普遍接受的作用之外,还能为最初被经典途径和凝集素途径引发的补体活化提供放大环路(Liszewski,M.K.和J.P.Atkinson,1993,载于Fundamental Immunology,第三版,由W.E.Paul编辑,Raven Press,Ltd.,New York;Schweinie,J.E.等,J.Clin.Invest.84:1821-1829,1989)。在这种替代途径介导的放大机制中,通过经典或凝集素补体级联活化而产生的C3转化酶(C4b2b)将C3切割成C3a和C3b,从而提供了能够参与形成C3bBb的C3b,C3bBb是替代途径C3转化酶。在MASP-2敲除血清中缺乏替代途径活化可能的解释是,由酵母聚糖、甘露聚糖以及其它推定的替代途径“激活因子”产生的初始的补体活化需要凝集素途径,而替代途径起着放大补体活化的关键作用。换句话说,替代途径是依赖凝集素和经典补体途径用于活化的前馈放大环路,而不是独立的线性级联。
补体级联并不是如以前设想的那样通过三条截然不同的途径(经典途径、替代途径和凝集素途径)被激活,我们的结果表明,将补体看成由两个主要系统组成更为准确,大致上相当于补体免疫防御系统的先天性(凝集素)和获得性(经典)的两翼。凝集素(MBP、M-纤维胶凝蛋白、H-纤维胶凝蛋白和L-纤维胶凝蛋白)是引发先天性补体系统的特异性识别分子,该系统包括凝集素途径和相关的替代途径放大环路。C1q是引发获得性补体系统的特异性识别分子,该系统包括经典途径和相关的替代途径放大环路。我们将这两个主要的补体活化系统分别称为凝集素依赖性补体系统和C1q依赖性补体系统。
除了在免疫防御中的基本作用外,补体系统是造成许多临床疾病的组织损伤的原因。因此,迫切需要开发治疗上有效的补体抑制剂以抑制这些不利作用。如果认识到补体是由两个主要的补体活化系统组成的,则就能够理解我们非常需要的是仅仅特异性抑制引起特定病理的补体活化系统而又不完全停止补体的免疫防御能力。例如,在其中补体活化主要由凝集素依赖性补体系统介导的疾病中,仅特异性抑制该系统将是有利的。这将保持C1q依赖性补体活化系统的完整性以处理免疫复合物加工以及帮助宿主防御感染。
在特异性抑制凝集素依赖性补体系统的治疗药物的研发中,优选的作为靶标的蛋白成分是MASP-2。在凝集素依赖性补体系统的所有蛋白成分(MBL、H-纤维胶凝蛋白、M-纤维胶凝蛋白、L-纤维胶凝蛋白、MASP-2、C2-C9、B因子、D因子和备解素)中,只有MASP-2是凝集素依赖性补体系统所独有并且是这个系统起作用所必需的。凝集素(MBL、H-纤维胶凝蛋白、M-纤维胶凝蛋白和L-纤维胶凝蛋白)也是凝集素依赖性补体系统中独有的成分。然而,这些凝集素成分任一种的丧失并不一定抑制系统活化,这是由于凝集素冗余所致。为了保证抑制凝集素依赖性补体活化系统,抑制所有4种凝集素可能是必要的。此外,由于还已知MBL和纤维胶凝蛋白具有不依赖于补体的调理活性,因此抑制凝集素功能将导致丧失这种有利的抗感染的宿主防御机制。相反,如果MASP-2是抑制靶标的话,这种不依赖于补体的凝集素调理活性会保持完整。MASP-2作为抑制凝集素依赖性补体活化系统的治疗靶的额外好处是,MASP-2的血浆浓度是所有补体蛋白中最低的(约500ng/ml);因此,为得到完全抑制可能只需要相对低浓度的高亲和性MASP-2抑制剂(Moller-Kristensen,M.等,J.Immunol Methods 282:159-167,2003)。
III.MASP-2在各种疾病和病症中的作用以及使用MASP-2抑制剂的治疗方法
缺血再灌注损伤
当血流在长期缺血后恢复时会发生缺血再灌注损伤(I/R)。它是许多疾病的发病和死亡的共同原因。外科手术患者在主动脉瘤修复术、心肺分流术、与例如器官移植(如心、肺、肝、肾)和/或四肢/指趾再植相关的血管吻合术、中风、心肌梗塞、休克和/或外科手术后的血液动力复苏之后易受其影响。患有动脉粥样硬化疾病的患者易发生心肌梗塞、中风以及血栓诱发的肠和下肢缺血。创伤患者常常遭受四肢一过性缺血。此外,任何原因的大量血液损失都导致全身I/R反应。
I/R损伤的病理生理学是复杂的,至少两个主要因素是造成这个过程的原因:补体活化和嗜中性粒细胞刺激和与之相伴的氧自由基介导的损伤。在I/R损伤中,补体活化最早记载于30多年前的心肌梗塞期间,并引起了关于补体系统对I/R组织损伤的影响的大量研究(Hill,J.H.等,J.Exp.Med.133:885-900,1971)。目前,不断积累的证据表明,补体是I/R损伤中的关键介质。在几种I/R动物模型中,补体抑制已经在限制损伤方面获得了成功。在早期的研究中,输注眼镜蛇毒因子之后使得C3耗尽,据报道该C3耗尽在肾脏和心脏的I/R期间是有利的(Maroko,P.R.等,1978,J.Clin Invest.61:661-670,1978;Stein,S.H.等,Miner Electrolyte Metab.11:256-61,1985)。然而,可溶形式的补体受体1(sCR1)是用来预防心肌I/R损伤的第一种补体特异性抑制剂(Weisman,H.F.等,Science 249:146-51,1990)。心肌I/R期间的sCR1治疗减少了梗塞形成,所述梗塞形成与沿冠状内皮沉积的C5b-9复合物的减少和再灌注后的白细胞浸润减少有关。
在实验性心肌I/R中,在再灌注之前给予C1酯酶抑制剂(C1INH)防止了C1q的沉积并显著减少了心肌坏死面积(Buerke,M.等,1995,Circulation 91:393-402,1995)。遗传上缺失C3的动物在骨骼肌或肠缺血之后,其局部组织坏死较少(Weiser,M.R.等,J.Exp.Med.183:2343-48,1996)。
膜攻击复合物是补体导向的损伤的最后媒介物,C5缺陷型动物的研究表明,I/R损伤模型中局部损伤和较远部位损伤减少(Austen,W.G.Jr.等,Surgery 126:343-48,1999)。已经证实在鼠肠再灌注开始前后分别给予补体活化抑制剂即可溶性Crry(补体受体相关基因Y)时对于抗损伤是有效的(Rehrig,S.等,J.Immunol.167:5921-27,2001)。在骨骼肌缺血模型中,在再灌注开始之后给予时,用可溶性补体受体1(sCR1)同样减少肌肉损伤(Kyriakides,C.等,Am.J.Physiol Cell Physiol.281:C244-30,2001)。在心肌I/R的猪模型中,在再灌注之前用针对过敏毒素C5a的单克隆抗体(“MoAb”)处理的动物显示,梗塞形成减少(Amsterdam,E.A.等,Am.J.Physiol.Heart Circ.Physiol.268:H448-57,1995)。用C5MoAb处理的大鼠证实,心肌中梗塞大小、嗜中性粒细胞浸润和凋亡减少(Vakeva,A.等,Circulation 97:2259-67,1998)。这些实验结果使I/R损伤发病机制中补体活化的重要性得到突出。
尚不清楚主要是哪条补体途径(经典、凝集素或替代途径)参与了I/R损伤中的补体活化。Weiser等人根据血管通透性显著降低表明C3和C4敲除小鼠受到保护而免受I/R损伤,从而证实了凝集素途径和/或经典途径在骨骼I/R期间的重要作用(Weiser,M.R.等,J.Exp.Med.183:2343-48,1996)。相反,用C4敲除小鼠进行的肾I/R实验表明没有显著的组织保护作用,而C3、C5和C6敲除小鼠则受到保护而免受损伤,这表明肾I/R损伤期间的补体活化是通过替代途径而发生的(Zhou,W.等,J.Clin.Invest.105:1363-71,2000)。Stahl等人最近采用D因子缺陷型小鼠,提供了关于替代途径在小鼠肠I/R中的重要作用的证据(Stahl,G.等,Am.J.Pathol.162:449-55,2003)。相反,Williams等人表明C4和IgM(Rag1-/-)缺陷型小鼠中C3的器官染色减少并保护器官免受损伤,从而提出小鼠肠I/R损伤开始时经典途径的重要作用(Williams,J.P.等,J.Appl.Physiol.86:938-42,1999)。
在心肌I/R模型中,用抗大鼠甘露聚糖结合凝集素(MBL)的单克隆抗体处理大鼠导致缺血后再灌注损伤减轻(Jordan,J.E.等,Circulation 104:1413-18,2001)。MBL抗体还在氧化应激之后,体外减少补体沉积在内皮细胞上,这表明了凝集素途径在心肌I/R损伤中的作用(Collard,C.D.等,Am.J.Pathol.156:1549-56,2000)。也有证据表明在一些器官中,I/R损伤可能由称为天然抗体的特定类别的IgM以及经典途径的活化所介导(Fleming,S.D.等,J.Immunol.169:2126-33,2002;Reid,R.R.等,J.Immunol.169:5433-40,2002)。
已经开发出几种补体活化抑制剂作为潜在治疗药物,来预防由心肌I/R并发症导致的发病率和死亡率。这些抑制剂中有两种即sCR1(TP10)和人源化抗C5scFv(培克珠单抗(Pexelizumab))已经完成了II期临床试验。培克珠单抗还完成了III期临床试验。尽管TP10在早期的I/II期试验中具有良好的耐受性,并且对患者有益,但是2002年2月结束的II期试验结果并没有达到初步终点。然而,来自正在进行心内直视手术的高危人群男性患者的数据的亚组分析证实,死亡率和梗塞大小显著降低。在COMA和COMPLY II期试验中,给予人源化抗C5scFv降低了与急性心肌梗塞有关的总体患者死亡率,但是未能达到初步终点(Mahaffey,K.W.等,Circulation 108:1176-83,2003)。最近发表了来自最新的抗C5scFv III期临床试验(PRIMO-CABG)的结果,其用于改进冠状动脉分流术后由手术产生的结果。尽管没有达到此项研究的初步终点,但是此研究证实,术后患者的发病率和死亡率整体下降。
Walsh博士及其同事证实,缺乏MBL、从而缺乏MBL依赖性凝集素途径活化但经典补体途径却充分活化的小鼠受到保护而不受心脏再灌注损伤,结果保护了心脏功能(Walsh等,J.Immunol.175:541-46,2005)。值得注意的是,缺乏经典补体途径识别成分C1q、但是具有完整MBL补体途径的小鼠,无法受到保护而不受损伤。这些结果表明凝集素途径在心肌缺血再灌注损伤的发病机制中具有主要作用。
已知补体活化在胃肠缺血再灌注(I/R)有关的组织损伤中起着重要作用。Hart及其同事采用GI/R鼠模型的最新研究指出,遗传上缺失MBL的小鼠在胃肠I/R后受到保护而不受肠损伤(Hart等,J.Immunol.174:6373-80,2005)。在胃肠I/R后,与未处理MBL缺陷型小鼠相比,在MBL缺陷型小鼠中加入重组MBL显著增加损伤。相比之下,遗传上缺失经典途径识别成分C1q的小鼠,在胃肠I/R之后未受到组织损伤的保护。
肾I/R是急性肾衰竭的重要原因。补体系统似乎基本上参与了肾I/R损伤。在最近的一项研究中,de Vries及其同事报道了在实验性肾I/R损伤以及临床肾I/R损伤进程中凝集素途径被激活(de Vries等,Am.J.Path.165:1677-88,2004)。此外,在肾I/R进程中,凝集素途径发生在补体C3、C6和C9沉积之前,并且与补体C3、C6和C9沉积共定位。这些结果表明凝集素途径的补体活化参与了肾I/R损伤。
因此,本发明的一个方面涉及通过应用药物载体中的治疗有效量的MASP-2抑制剂来治疗经历缺血再灌注的受试者从而治疗缺血再灌注损伤。可通过动脉内、静脉内、颅内、肌内、皮下或其它胃肠外给药,给予受试者MASP-2抑制剂,对于非肽能抑制剂可口服给予,最适合经动脉内或静脉内给药。本发明的MASP-2抑制剂组合物适于在缺血再灌注事件后立即或尽快开始给予。在受控环境下发生再灌注的情况下(例如在主动脉瘤修复术、器官移植或断肢或断指趾或者受伤四肢或指趾的复置术后),可以在再灌注之前和/或期间和/或之后给予MASP-2抑制剂。为达到最佳治疗效果,可按照医师规定定期重复给药。
动脉粥样硬化
有相当多的证据表明,补体活化参与人体动脉粥样化的形成。多项研究令人信服地表明,尽管在正常动脉中没有发生显著的补体活化,但是补体在动脉粥样硬化病变中被大量活化,在易受损和破裂的斑块中尤其突出。在人动脉粥样化中,经常发现末端补体途径的成分(Niculescu,F.等,Mol.Immunol.36:949-55.10-12,1999;Rus,H.G.等,Immunol.Lett.20:305-310,1989;Torzewski,M.等,Arterioscler.Thromb.Vasc.Biol.18:369-378,1998)。还发现在动脉病变中有C3和C4沉积(Hansson,G.K.等,Acta Pathol.Microbiol.Immunol.Scand.(A)92:429-35,1984)。C5b-9沉积的程度与损伤的严重性有关(Vlaicu,R.等,Atherosclerosis57:163-77,1985)。补体iC3b、而不是C5b-9沉积在破裂和易损斑块上的情况特别严重,这表明补体活化可能是急性冠状动脉综合征的一个因素(Taskinen S.等,Biochem.J.367:403-12,2002)。在兔实验性动脉粥样化中发现了补体活化先于病变发生前(Seifer,P.S.等,Lab Invest.60:747-54,1989)。
在动脉粥样硬化病变中,补体通过经典途径和替代途径被激活,但是至今还几乎没有补体活化是通过凝集素途径的证据。动脉壁的几个成分可能引发补体活化。可能通过与酶降解LDL结合的C反应蛋白(CRP)来激活补体的经典途径(Bhakdi,S.等,Arterioscler.Thromb.Vasc.Biol.19:2348-54,1999)。与此观点一致的发现是,末端补体蛋白与早期人病变内膜中的CRP共定位(Torzewski,J.等,Arterioscler.Thromb.Vasc.Biol.18:1386-92,1998)。同样地,对病变内的氧化LDL特异性的免疫球蛋白IgM或IgG抗体可能激活经典途径(Witztum,J.L.,Lancet 344:793-95,1994)。从人动脉粥样硬化病变中分离的脂质具有高含量的未酯化的胆固醇,并且能够激活替代途径(Seifert P.S.等,J.Exp.Med.172:547-57,1990)。常常与动脉粥样硬化病变相关的革兰氏阴性菌肺炎衣原体(Chlamydia pneumoniae)也可激活补体替代途径(Campbell L.A.等,J.Infect.Dis.172:585-8,1995)。动脉粥样硬化病变中存在的其它潜在的补体活化因子包括胆固醇结晶和细胞碎片,这两者都可激活替代途径(Seifert,P.S.等,Mol.Immunol.24:1303-08,1987)。
已知补体活化的副产物具有许多能够影响动脉粥样硬化病变发展的生物学性质。局部补体活化可能诱导细胞裂解并且产生至少一些在晚期病变坏死核心中发现的细胞碎片(Niculescu,F.等,Mol.Immunol.36:949-55.10-12,1999)。亚裂解补体活化可能是造成平滑肌细胞增殖以及动脉粥样化形成期间单核细胞浸润到动脉内膜上的重要因素(Torzewski J.等,Arterioscler.Thromb.Vasc.Biol.18:673-77,1996)。补体的持续活化可能是有害的,因为它可能引发炎症并使炎症持续。除了血浆补体成分的浸润外,动脉细胞表达了补体蛋白信使RNA,各种补体成分的表达在动脉粥样硬化病变中被上调(Yasojima,K.等,Arterioscler.Thromb.Vasc.Biol.21:1214-19,2001)。
已经报道的关于补体蛋白缺乏对动脉粥样化形成的影响的研究数量有限。实验动物模型中的结果也不一致。在大鼠中,通过有毒剂量的维生素D诱发的动脉粥样硬化样病变的形成在缺乏补体的动物体内减少(Geertinger P.等,Acta.Pathol.Microbiol.Scand.(A)78:284-88,1970)。此外,在胆固醇喂养兔中,通过遗传性C6缺乏症(Geertinger,P.等,Artery 1:177-84,1977;Schmiedt,W.等,Arterioscl.Thromb.Vasc.Biol.18:1790-1795,1998)或者通过抗补体药物K-76COONa(Saito,E.等,J.Drug Dev.3:147-54,1990)引起的补体抑制,抑制了动脉粥样硬化的发展而又不影响血清胆固醇水平。相反,最近的研究报道了脱脂载脂蛋白E(ApoE)缺陷型小鼠中,C5缺乏不减缓动脉粥样硬化病变的发展(Patel,S.等,Biochem.Biophys.Res.Commun.286:164-70,2001)。然而在另一项研究中,在存在或不存在下C3缺乏的情况下,对LDLR缺陷型(ldlr-)小鼠的动脉粥样硬化病变的发展进行了评价(Buono,C.等,Circulation 105:3025-31,2002)。他们发现动脉粥样化到动脉粥样硬化样病变的成熟部分取决于完整补体系统的存在。
因此,本发明的一个方面涉及通过应用药物载体中的治疗有效量的MASP-2抑制剂来治疗患有或易患动脉粥样硬化的受试者从而治疗或预防动脉粥样硬化。可以通过动脉内、静脉内、鞘内、颅内、肌内、皮下或其它胃肠外给药而给予受试者MASP-2抑制剂,对于非肽能抑制剂可口服给予。可以在受试者诊断出动脉粥样硬化后开始给予或者在发生该病的高危受试者中预防性地给予MASP-2抑制剂组合物。为达到最佳治疗效果,可按照医师规定定期重复给药。
其它血管疾病和病症
内皮很大程度上暴露于免疫系统,特别容易受到存在于血浆中的补体蛋白的攻击。研究表明补体介导的血管损伤是心血管系统中若干疾病的病理生理学原因,所述疾病包括动脉粥样硬化(Seifert,P.S.等,Atherosclerosis 73:91-104,1988)、缺血再灌注损伤(Weisman,H.F.,Science249:146-51,1990)和心肌梗塞(Tada,T.等,Virchows Arch 430:327-332,1997)。有证据表明,补体活化可能扩展到其它血管病中。
例如,有证据表明,补体活化是多种形式的血管炎的发病机制的原因之一,包括亨诺赫–舍恩莱因紫癜肾炎、与系统性红斑狼疮相关的血管炎、与类风湿性关节炎相关的血管炎(亦称恶性类风湿性关节炎)、免疫复合物型血管炎和无脉症。亨诺赫–舍恩莱因紫癜肾炎是具有免疫发病机制的小血管系统性血管炎的一种形式,其中补体通过凝集素途径活化导致C5b-9诱导的内皮损伤被认为是一个重要的机制(Kawana,S.等,Arch.Dermatol.Res.282:183-7,1990;Endo,M.等,Am J.Kidney Dis.35:401-7,2000)。系统性红斑狼疮(SLE)是影响多种器官的全身性自身免疫性疾病的实例,包括皮肤、肾、关节、浆膜表面和中枢神经系统,常常与严重血管炎有关。在患有活动性SLE患者的血清中存在IgG抗内皮抗体和能够结合内皮细胞的IgG复合物,并且在患有SLE血管炎患者的血管壁上发现了IgG免疫复合物和补体的沉积(Cines,D.B.等,J.Clin.Invest.73:611-25,1984)。与血管炎有关的类风湿性关节炎(亦称恶性类风湿性关节炎)(Tomooka,K.,Fukuoka Igaku Zasshi 80:456-66,1989)、免疫复合物型血管炎、与甲型肝炎有关的血管炎、破白细胞性血管炎以及被称为无脉症的动脉炎,构成了另一多型组别的人类疾病,其中针对内皮细胞和其它细胞类型的补体依赖的细胞毒性起着已证实的作用(Tripathy,N.K.等,J.Rheumatol.28:805-8,2001)。
还有证据表明了补体活化在扩张型心肌病中起作用。扩张型心肌病是以心脏肥大和心脏收缩功能受损为特征的综合征。最新数据表明,心肌中正在发生的炎症可能是疾病发生的原因。已知补体的末端膜攻击复合物C5b-9与免疫球蛋白沉积和TNF-α的心肌表达显著相关。在28名患有扩张型心肌病患者的心肌活检样品中,证实了C5b-9的心肌积累,这就表明心肌中慢性免疫球蛋白介导的补体活化可能是部分促成扩张型心肌病进程的原因(Zwaka,T.P.等,Am.J.Pathol.161(2):449-57,2002)。
因此,本发明的一个方面涉及通过给予包含治疗有效量的MASP-2抑制剂在药物载体中的组合物来治疗血管病,所述血管病包括心血管病、脑血管病、外周(如肌肉骨骼)血管病、肾血管病和肠系膜/肠血管病。认为本发明适用的疾病包括但不限于:血管炎,包括亨诺赫-舍恩莱因紫癜肾炎、与系统性红斑狼疮相关的血管炎、与类风湿性关节炎相关的血管炎(亦称恶性类风湿性关节炎)、免疫复合物型血管炎和无脉症;扩张型心肌病;糖尿病性血管病;川崎病(动脉炎)和静脉气体栓塞(VGE)。同样,如果补体活化的发生是作为与心血管介入手术相关的内腔创伤和异物炎症反应的结果,则认为本发明的MASP-2抑制剂组合物还可以单独或与其它再狭窄抑制剂组合,用于抑制支架放置后再狭窄、动脉粥样硬化斑旋切术后再狭窄和/或经皮经腔冠状动脉成形术(PTCA)后再狭窄,所述其它再狭窄抑制剂公开于Demopulos的美国专利第6,492,332号。
可以通过动脉内、静脉内、肌内、鞘内、颅内、皮下或其它胃肠外给药给予受试者MASP-2,对于非肽能抑制剂可口服给予。为达到最佳治疗效果,可按照医师规定定期重复给药。对于抑制再狭窄,可以在安置支架或者动脉粥样硬化斑切除术或者血管成形手术之前和/或期间和/或之后给予MASP-2抑制剂组合物。或者,可将MASP-2抑制剂组合物涂敷在支架上或掺入支架中。
胃肠疾病
溃疡性结肠炎和克罗恩病是炎性肠病(IBD)类别下的慢性肠炎疾病。IBD的特征是未知起因自发发生的慢性复发性炎症。尽管在人体和实验动物两者中对该病都进行了广泛深入研究,但是确切的病理学机制仍有待阐明。然而,有研究认为补体系统在IBD患者中被激活,而且认为补体系统在疾病发病机制中发挥作用(Kolios,G.等,Hepato-Gastroenterology 45:1601-9,1998;Elmgreen,J.,Dan.Med.Bull.33:222,1986)。
研究表明,在IBD患者的表层上皮细胞内腔面(luminal face)以及粘膜肌层和粘膜下层血管中发现了C3b和其它活化的补体产物(Halstensen,T.S.等,Immunol.Res.10:485-92,1991;Halstensen,T.S.等,Gastroenterology 98:1264,1990)。此外,在炎性肠病中发现了特有的多形核细胞浸润,这通常是C5a产生的结果(Kohl,J.,Mol.Immunol.38:175,2001)。还有报道指出在小型临床研究中(Kitano,A.等,Dis.Colon Rectum 35:560,1992)以及在角叉菜聚糖诱发的结肠炎兔模型中(Kitano,A.等,Clin.Exp.Immunol.94:348-53,1993),多功能补体抑制剂K-76使溃疡性结肠炎的症状得到改善。
已表明,一种新的人C5a受体拮抗剂保护大鼠IBD模型免于疾病病理(Woodruff,T.M.等,J.Immunol.171:5514-20,2003)。将遗传上缺失衰变加速因子(DAF,一种膜补体调控蛋白)的小鼠用于IBD模型中,表明DAF缺乏导致明显更多的组织损伤,而且促炎细胞因子的产生增加(Lin,F.等,J.Immunol.172:3836-41,2004)。因此,补体调控在肠稳态调节中十分重要,可能是参与IBD发生的主要发病机制。
因此,本发明提供用于抑制患有炎性胃肠疾病的受试者的MASP-2依赖性补体活化的方法,该方法通过给予患有这些疾病的患者包含治疗有效量的MASP-2抑制剂在药物载体中的组合物实施,所述胃肠疾病包括但不限于胰腺炎、憩室炎和肠病,包括克罗恩病、溃疡性结肠炎和过敏性肠综合征。可通过动脉内、静脉内、肌内、皮下、鞘内、颅内或其它胃肠外给药来给予受试者MASP-2抑制剂,对于非肽能抑制剂可口服给予。可按照医师的规定适当地定期重复给药以控制待治疗疾病的症状。
肺部疾病
许多肺部炎性疾病的发病机制中涉及补体,所述疾病包括急性呼吸窘迫综合征(ARDS)(Ware,I.等,N.Engl.J.Med.342:1334-49,2000);输血相关性急性肺损伤(TRALI)(Seeger,W.等,Blood 76:1438-44,1990);缺血/再灌注急性肺损伤(Xiao,F.等,J.Appl.Physiol.82:1459-65,1997);慢性阻塞性肺病(COPD)(Marc,M.M.等,Am.J.Respir.Cell Mol.Biol.(印刷前的电子出版物),2004年3月23日);哮喘(Krug,N.等,Am.J.Respir.Crit.Care Med.164:1841-43,2001);韦格纳肉芽肿病(Kalluri,R.等,J.Am.Soc.Nephrol.8:1795-800,1997);和抗肾小球基底膜病(古德帕斯彻病)(Kondo,C.等,Clin.Exp.Immunol.124:323-9,2001)。
目前普遍接受的是,许多ARDS的病理生理学涉及炎症级联失调,这种炎症级联开始是作为对感染或其它刺激性事件的正常反应,但是最终引起了宿主显著的自体损伤(Stanley,T.P.,Emerging Therapeutic Targets 2:1-16,1998)。ARDS患者几乎全都显示大范围补体活化的证据(补体成分C3a和C5a的血浆水平升高),补体活化的程度与ARDS的发生和结果有关(Hammerschmidt,D.F.等,Lancet 1:947-49,1980;Solomkin,J.S.等,J.Surgery97:668-78,1985)。
各种实验数据和临床数据表明补体活化在ARDS病理生理学中的作用。在动物模型中,补体的系统活化导致急性肺损伤,其组织病理学与在人ARDS中发现的类似(Till,G.O.等,Am.J.Pathol.129:44-53,1987;Ward,P.A.,Am.J.Pathol.149:1081-86,1996)。通过一般性补体耗竭或者通过特异性抑制C5a从而抑制补体级联,为急性肺损伤动物模型提供了保护作用(Mulligan,M.S.等,J.Clin.Invest.98:503-512,1996)。在大鼠模型中,sCR1在补体介导的肺损伤和嗜中性粒细胞介导的肺损伤中具有保护作用(Mulligan,M.S.、Yeh等,J.Immunol.148:1479-85,1992)。此外,所有补体成分实际上都可通过II型肺泡细胞、肺泡巨噬细胞和肺成纤维细胞在肺中局部产生(Hetland,G.等,Scand.J.Immunol.24:603-8,1986;Rothman,B.I.等,J.Immunol.145:592-98,1990)。因此,补体级联定位准确从而显著引起肺部炎症,因此在ARDS中导致肺损伤。
哮喘本质上是炎性疾病。变应性哮喘最重要的特征包括对各种特异性和非特异性刺激的气道高反应性、气道粘液过度产生、肺部嗜酸粒细胞增多以及血清IgE浓度升高。尽管哮喘的起因是多因子的,不过一般认为,它是在遗传易感个体中作为对普通环境抗原不当免疫应答的结果而产生的。文献中大量记载了人哮喘性肺中补体系统被高度激活这一事实(Humbles,A.A.等,Nature 406:998-01,2002;van de Graf,E.A.等,J.Immunol.Methods147:241-50,1992)。此外,得自动物模型和人体的最新的数据为补体活化是导致疾病发病机制的重要机制提供了证据(Karp,C.L.等,Nat.Immunol.1:221-26,2000;Bautsch,W.等,J.Immunol.165:5401-5,2000;Drouin,S.M.等,J.Immunol.169:5926-33,2 002;Walters,D.M.等,Am.J.Respir.Cell Mol.Biol.27:413-18,2002)。使用慢性真菌性哮喘鼠模型的研究支持凝集素途径在哮喘中的作用。在该哮喘模型中,甘露聚糖结合凝集素的遗传缺陷型小鼠与正常动物相比,气道高反应性发生改变(Hogaboam,C.M.等,J.Leukoc.Biol.75:805-14,2004)。
在哮喘中,可通过几种途径激活补体,包括:(a)作为变应原-抗体复合物形成的结果通过经典途径而被活化;(b)变应原表面上的替代途径活化;(c)凝集素途径通过变应原上结合糖类结构而被活化;以及(d)通过从炎性细胞释放出的蛋白酶切割C3和C5。尽管在哮喘中,还有很多有关由补体所起的复合物作用的方面尚待研究,但是鉴定变应性哮喘发生中所涉及的补体活化途径,可以为开发这种日趋重要的疾病的新治疗策略提供方向。
使用动物模型的多项研究证实,C3及其裂解产物C3a在变应型表型发生中的关键作用。Drouin及其同事在卵清蛋白(OVA)/烟曲霉(Aspergillus fumigatus)哮喘模型中使用了C3缺陷型小鼠(Drouin等,J.Immunol.167:4141-45,2001)。他们发现,C3缺陷型小鼠当用变应原攻击时,与匹配的野生型对照小鼠相比,AHR和肺嗜酸粒细胞增多的减少。此外,这些C3缺陷型小鼠显著地减少产生IL-4的细胞的数目,并且削弱Ag特异型IgE和IgG1应答。Taube及其同事在OVA哮喘模型中,通过使用可溶性重组形式的小鼠补体受体Crry,在C3和C4水平阻断补体活化方面,得到了类似的结果(Taube等,Am.J.Respir.Crit.Care Med.168:1333-41,2003)。Humbles及其同事在小鼠中剔除了C3aR以检查C3a在嗜酸性粒细胞功能中的作用(Humbles等,Nature406:998-1001,2000)。他们使用OVA哮喘模型,观察到对于气溶胶化乙酰甲基胆碱几乎得到完全保护而不发生AHR。Drouin及其同事(2002)曾在OVA/烟曲霉哮喘模型中使用C3aR缺陷型小鼠,并且证实变应性反应的减弱与肺中AHR减轻、嗜酸性粒细胞募集、TH2细胞因子产生和粘液分泌减少以及Ag特异型IgE和IgG1应答减弱的C3缺陷型动物非常相似(Drouin等,J.Immunol.169:5926-33,2002)。Bautsch及其同事使用天然缺失C3aR的豚鼠品系进行了研究(Bautsch等,J.Immunol.165:5401-05,2000)。他们使用OVA变应性哮喘模型,观察到在抗原攻击后受到显著保护而免于气道支气管收缩。
近来多项使用动物模型的研究证实了C5及其裂解产物C5a在变应型表型发生中的关键作用。Abe及其同事报道了C5aR活化与气道炎症、细胞因子产生和气道反应性相关的证据(Abe等,J.Immunol.167:4651-60,2001)。在他们的研究中,通过可溶性CR1、(补体活化抑制剂)futhan或合成的六肽C5a拮抗剂来抑制补体活化,阻断了炎症反应和对乙酰甲基胆碱的气道反应性。在采用阻断抗C5单克隆抗体的研究中,Peng及其同事发现在OVA哮喘模型中,C5活化基本上是造成气道炎症和AHR的原因(Peng等,J.Clin.Invest.115:1590-1600,2005)。Baelder及其同事同样也报道了C5aR的阻断基本上减轻了烟曲霉哮喘模型的AHR(Baelder等,J.Immunol.174:783-89,2005)。此外,C3aR和C5aR两者的阻断显著减轻了气道炎症,如BAL中嗜中性粒细胞和嗜酸性粒细胞数目减少所证实的一样。
尽管以上罗列的研究突出了补体因子C3和C5及其裂解产物在实验性变应性哮喘发病机制中的重要性,但是这些研究没有提供有关三条补体活化途径中每一条所起作用的资料,因为C3和C5是所有三条活化途径共有的。然而,Hogaboam及其同事的最新研究表明,凝集素途径可能在哮喘发病机制中具有主要作用(Hogaboam等,J.Leukocytye Biol.75:805-814,2004)。这些研究使用甘露聚糖结合凝集素-A(MBL-A,一种起着凝集素补体途径活化识别成分的作用的糖结合蛋白)遗传缺陷型小鼠。在慢性真菌性哮喘模型中,在用烟曲霉分生孢子i.t.攻击后的第4天和第48天,检查MBL-A(+/+)和MBL-A(-/-)烟曲霉敏化小鼠。与敏化MBL-A(+/+)组相比,敏化MBL-A(-/-)小鼠在分生孢子攻击后的两个时间段上,AHR都显著减缓。他们发现与野生型动物组相比,烟曲霉敏化MBL-A(-/-)小鼠肺中的TH2细胞因子水平(IL-4、IL-5和IL-13)在分生孢子攻击后第4天显著降低。他们的结果表明,在慢性真菌性哮喘期间MBL-A和凝集素途径在AHR的发生和持续中具有主要作用。
最新的特异性MBL多态性与哮喘发生之间相关性的临床研究的结果进一步为凝集素途径可能在该病中起着重要病理作用提供了证据(Kaur等,Clin.Experimental Immunol.143:414-19,2006)。个体间MBL的血浆浓度变化很大,这主要归结于MBL基因内的遗传多态性。他们发现携带至少一个拷贝的使MBL表达上调2-4倍的特异性MBL多态性的个体,发生支气管哮喘的风险增加了几乎5倍。在携带该MBL多态性的支气管哮喘患者中,这也是疾病严重性增强的标志。
因此,本发明的一方面提供用于治疗肺部疾病的方法,该方法通过将包含治疗有效量的MASP-2抑制剂在药物载体中的组合物给予患有肺部疾病的受试者实施,所述疾病包括但不限于急性呼吸窘迫综合征、输血相关性急性肺损伤、缺血/再灌注急性肺损伤、慢性阻塞性肺病、哮喘、韦格纳肉芽肿病、抗肾小球基底膜病(古德帕斯彻病)、胎粪吸入综合征、闭塞性细支气管炎综合征、特发性肺纤维化、烧伤继发性急性肺损伤、非心源性肺水肿、输血相关的呼吸抑制和肺气肿。可以全身性给予受试者MASP-2抑制剂,例如通过动脉内、静脉内、肌内、吸入、经鼻、皮下或者其它胃肠外给药,或者对于非肽能药物可通过口服给药。MASP-2抑制剂组合物可以与一种或多种另外的治疗药物组合,另外的治疗药物包括抗炎药、抗组胺药、皮质类固醇或者抗微生物药。可按照医师规定重复给药直到疾病消退。
体外循环
有许多医学手术在其实施期间要从患者循环系统转移血液(体外循环系统或ECC)。这些手术包括血液透析法、血浆除去法、白细胞除去法、体外膜式氧合(ECMO)、肝素诱导的体外膜式氧合LDL沉淀(HELP)和心肺分流术(CPB)。这些手术将血液或血液产物暴露给可能改变正常细胞功能和止血的外源表面。在最早的研究中,Craddock等人鉴定出补体活化是血液透析期间粒细胞减少的可能原因(Craddock,P.R.等,N.Engl.J.Med.296:769-74,1977)。从1977年到现在,无数研究结果表明,进行血液透析或CPB的患者所经受的许多不良事件是由补体系统活化引起的(Chenoweth,D.E.,Ann.N.Y.Acad.Sci.516:306-313,1987;Hugli,TE,Complement 3:111-127,1986;Cheung,A.K.,J.Am.Soc.Nephrol.1:150-161,1990;Johnson,R.J.,Nephrol.Dial.Transplant 9:36-45 1994)。例如,已经表明补体活化潜力是确定血液透析器与肾功能恢复、易感染性、肺功能障碍、肾衰竭患者的发病率和存活率方面的生物相容性的重要标准(Hakim,R.M.,Kidney Int.44:484-4946,1993)。
在很大程度上认为,通过血液透析膜的补体活化是通过替代途径机制而发生的,这是由于C4a产生很少(Kirklin,J.K.等,J.Thorac.Cardiovasc.Surg.86:845-57,1983;Vallhonrat,H.等,ASAIO J.45:113-4,1999),但是最新研究提出可能还包括经典途径(Wachtfogel,Y.T.等,Blood 73:468-471,1989)。然而,对启动和控制人工表面(包括生物医药用聚合物)上补体活化的因素了解得仍然不够。例如,已将用于血液透析中的铜仿膜(Cuprophan membrane)归类为非常有效的补体活化因子。尽管不希望受理论束缚,但是本发明的发明人还是推测这也许可部分通过其多糖性质来解释。在本专利中所鉴定的MASP-2依赖性补体活化系统提供了一种机制,由此凝集素途径的活化便引发了替代途径活化。
在CPB期间进行ECC的患者遭受全身性炎症反应,这部分是由血液暴露于体外环路的人工表面而引起的,但也是由不依赖于表面的因素象外科创伤和缺血再灌注损伤引起的(Butler,J.等,Ann.Thorac.Surg.55:552-9,1993;Edmunds,L.H.,Ann.Thorac.Surg.66(增刊):S12-6,1998;Asimakopoulos,G.,Perfusion 14:269-77,1999)。CPB引发的炎症反应可能导致术后并发症,一般称为“灌注后综合征”。在这些术后事件中有认知缺陷(Fitch,J.等,Circulation 100(25):2499-2506,1999)、呼吸衰竭、出血性疾病、肾功能障碍,最严重的情况下为多器官衰竭(Wan,S.等,Chest 112:676-692,1997)。与没有CPB但是有相应程度外科创伤的手术相比,有CPB的冠状旁路手术导致了更多的补体活化(E.Fosse,1987)。因此,这些CPB相关问题主要的疑似因素是旁路手术期间的补体活化不当(Chenoweth,K.等,N.Engl.J.Med.304:497-503,1981;Haslam.P等,Anaesthesia 25:22-26,1980;J.K.Kirklin等,J.Thorac.Cardiovasc.Surg.86:845-857,1983;Moore,F.D.等,Ann.Surg 208:95-103,1988;Steinberg.J等,J.Thorac.Cardiovasc.Surg 106:1901-1918,1993)。在CPB环路中,替代补体途径在补体活化中发挥了主要作用,这是血液与CPB环路人工表面之间相互作用的结果(Kirklin,J.K.等,J.Thorac.Cardiovasc.Surg.,86:845-57,1983;Kirklin,J.K.等,Ann.Thorac.Surg.41:193-199,1986;Vallhonrat H.等,ASAIO J.45:113-4,1999)。然而,也有证据表明CPB期间经典补体途径被激活(Wachtfogel,Y.T.等,Blood 73:468-471,1989)。
主要的炎症物质是在补体系统活化后产生的,包括过敏毒素C3a和C5a、调理素C3b和膜攻击复合物C5b-9。C3a和C5a是嗜中性粒细胞、单核细胞和血小板的有效刺激物(Haeffner-Cavaillon,N.等,J.Immunol.,139:794-9,1987;Fletcher,M.P.等,Am.J.Physiol.265:H1750-61,1993;Rinder,C.S.等,J.Clin.Invest.96:1564-72,1995;Rinder,C.S.等,Circulation 100:553-8,1999)。这些细胞的活化导致促炎细胞因子(IL-1、IL-6、IL-8、TNFα)、氧自由基和蛋白酶的释放(Schindler,R.等,Blood 76:1631-8,1990;Cruickshank,A.M.等,Clin Sci.(Lond)79:161-5,1990;Kawamura,T.等,Can.J.Anaesth.40:1016-21,1993;Steinberg,J.B.等,J.Thorac.Cardiovasc.Surg.106:1008-1,1993;Finn,A.等,J.Thorac.Cardiovasc.Surg.105:234-41,1993;Ashraf,S.S.等,J.Cardiothorac.Vasc.Anesth.11:718-22,1997)。已经表明C5a使多形核细胞(PMN)中Mac-1的粘着分子CD11b和CD18上调,并诱导PMN脱粒以释放促炎酶(Rinder,C.等,Cardiovasc Pharmacol.27(增刊1):S6-12,1996;Evangelista,V.等,Blood 93:876-85,1999;Kinkade,J.M.,Jr.等,Biochem.Biophys.Res.Commun.114:296-303,1983;Lamb,N.J.等,Crit.Care Med.27:1738-44,1999;Fujie,K.等,Eur.J.Pharmacol.374:117-25,1999)。C5b-9诱导血小板上的粘着分子P选择蛋白(CD62P)的表达(Rinde,C.S.等,J.Thorac.Cardiovasc.Surg.118:460-6,1999),而C5a和C5b-9两者都诱导内皮细胞上P选择蛋白的表面表达(Foreman,K.E.等,J.Clin.Invest.94:1147-55,1994)。这些粘着分子参与白细胞、血小板和内皮细胞之间的相互作用。粘着分子在活化内皮细胞上的表达是活化白细胞汇集的原因,然后介导组织炎症和损伤(Evangelista,V.,Blood 1999;Foreman,K.E.,J.Clin.Invest.1994;Lentsch,A.B.等,J.Pathol.190:343-8,2000)。正是这些补体活化产物对嗜中性粒细胞、单核细胞、血小板和其它循环细胞的作用才可能导致在CPB后出现各种问题。
正在研究若干补体抑制剂在CPB中的潜在应用。它们包括可溶性重组补体受体1(sCR1)(Chai,P.J.等,Circulation 101:541-6,2000)、人源化单链抗C5抗体(h5G1.1-scFv或培克珠单抗)(Fitch,J.C.K.等,Circulation 100:3499-506,1999)、人膜辅因子蛋白和人衰变加速因子的重组融合杂合体(CAB-2)(Rinde,C.S.等,Circulation 100:553-8,1999)、13个残基的C3结合环肽(Compstatin)(Nilsson,B.等,Blood 92:1661-7,1998)和抗D因子MoAb(Fung,M.等,J.Thoracic Cardiovasc.Surg.122:113-22,2001)。SCR1和CAB-2在C3和C5活化步骤抑制经典和替代补体途径。Compstatin在C3活化步骤抑制两条补体途径,而h5G1.1-scFv仅在C5活化步骤抑制两条补体途径。抗D因子MoAb在C3和C5活化步骤抑制替代途径。然而,这些补体抑制剂都不会特异性抑制本专利所鉴定的MASP-2依赖性补体活化系统。
已经报道了大有希望的III期临床研究的结果,研究人源化单链抗C5抗体(h5G1.1-scFv,培克珠单抗(pexelizu mab))在降低围手术期MI和冠状动脉旁路移植(CABG)手术死亡率中的功效和安全性(Verrie,E.D.等,JAMA 291:2319-27,2004)。与安慰剂相比,培克珠单抗(pexelizu mab)与已实施CABG手术的2746名患者中的最终死亡或MI混合风险的显著降低没有关联。然而,在进行包括或不包括瓣膜手术的CABG手术的所有3099名患者中,术后30天有统计学上的显著降低。由于培克珠单抗(pexelizu mab)在C5活化步骤抑制C5a和sC5b-9的产生,但是对其它两种有效的补体炎性物质C3a和调理素C3b的产生没有影响,亦知C3a和C3b是CPB引发的炎症反应的原因。
因此,本发明的一个方面涉及通过应用包含治疗有效量的MASP-2抑制剂在药物载体中的组合物来治疗进行体外循环手术的受试者,从而预防或治疗体外暴露所引发的炎症反应,所述受试者包括进行血液透析、血浆除去法、白细胞除去法、体外膜式氧合(ECMO)、肝素诱导的体外膜式氧合LDL沉淀(HELP)和心肺分流术(CPB)的患者。认为按照本发明方法的MASP-2抑制剂治疗可用于降低或预防有时由CPB手术引起的认知功能障碍。可在手术之前和/或手术之中和/或手术之后,通过例如动脉内、静脉内、肌内、皮下或其它胃肠外给药给予受试者MASP-2抑制剂。或者,可在体外循环期间将MASP-2抑制剂引入受试者血流中,例如通过将MASP-2抑制剂注射到血液通过或流过其进行循环的管或膜中,或者通过使血液与涂敷有MASP-2抑制剂的表面例如管、膜的内壁或其它表面如CPB装置相接触。
炎性关节炎和非炎性关节炎以及其它肌肉骨骼疾病
各种各样的风湿性疾病的发病机制中涉及补体系统的活化;包括类风湿性关节炎(Linton,S.M.等,Molec.Immunol.36:905-14,1999)、青少年类风湿性关节炎(Mollnes,T.E.等,Arthritis Rheum.29:1359-64,1986)、骨关节炎(Kemp,P.A.等,J.Clin.Lab.Immunol.37:147-62,1992)、系统性红斑狼疮(SLE)(Molina,H.,Current Opinion in Rheumatol.14:492-497,2002)、贝赫切特综合征(Behcet's syndrome)(Rumfeld,W.R.等,Br.J.Rheumatol.25:266-70,1986)和斯耶格伦综合征(Sjogren's syndrome)(Sanders,M.E.等,J.Immunol.138:2095-9,1987)。
很有说服力的证据表明,免疫复合物引发的补体活化是造成类风湿性关节炎(RA)组织损伤的主要病理机制。许多出版物记载了RA患者血浆中补体活化产物增加(Morgan,B.P.等,Clin.Exp.Immunol,73:473-478,1988;Auda,G.等,Rheumatol.Int.10:185-189,1990;Rumfeld,W.R.等,Br.J.Rheumatol.25:266-270,1986)。还在发炎的风湿性关节中发现了如C3a、C5a和sC5b-9的补体活化产物,并且已经确定补体活化程度和RA严重性之间正相关(Makinde,V.A.等,Ann.Rheum.Dis.48:302-306,1989;Brodeu,J.P.等,Arthritis Rheumatism34:1531-1537,1991)。在成人和青少年类风湿性关节炎中,替代途径补体活化产物Bb的血清和滑液水平比C4d(经典途径活化的标记)的高,表明补体活化主要是由替代途径介导的(El-Ghobarey,A.F.等,J.Rheumatology 7:453-460,1980;Agarwal,A.等,Rheumatology39:189-192,2000)。补体活化产物可直接损伤组织(通过C5b-9)或者经过敏毒素C3a和C5a募集炎性细胞而间接介导炎症。
实验性关节炎的动物模型已被广泛用来研究补体在RA发病机制中的作用。在RA动物模型中,通过眼镜蛇毒因子耗尽补体防止了关节炎的发生(Morgan,K.等,Arthritis Rheumat.24:1356-1362,1981;Van Lent,P.L.等,Am.J.Pathol.140:1451-1461,1992)。将可溶形式的补体受体1(sCR1,一种补体抑制剂)注射到关节内,抑制了RA大鼠模型中的炎症(Goodfellow,R.M.等,Clin.Exp.Immunol.110:45-52,1997)。此外,sCR1抑制大鼠胶原诱发性关节炎的发生和进程(Goodfellow,R.M.等,Clin Exp.Immunol.119:210-216,2000)。可溶性CR1在替代途径和经典途径的C3和C5活化步骤抑制经典和替代补体途径,从而抑制C3a、C5a和sC5b-9的产生。
1970年代后期,认识到用异种II型胶原(CII;人关节软骨的主要胶原成分)免疫啮齿动物导致十分类似于人RA的自身免疫性关节炎(胶原诱发性关节炎,或者CIA)的发生(Courtenay,J.S.等,Nature 283:666-68,1980;Banda等,J.of Immunol.171:2109-2115(2003))。在易感动物中,自身免疫应答涉及各因素的复杂组合,包括特定的主要组织相容性复合物(MHC)分子、细胞因子和CII特异性的B细胞和T细胞应答(有关综述见Myers,L.K.等,Life Sciences 61:1861-78,1997)。几乎40%的近交系小鼠的补体成分C5完全缺乏的观察结果(Cinade,B.等,J.Exp.Med.120:897-902,1964),为通过对C5缺陷型与C5充足型品系之间进行CIA比较从而研究补体在该关节炎模型中的作用提供了间接的机会。这些研究结果表明,足够的C5绝对是CIA发生所必需的(Watson等,1987;Wang,Y.等,J.Immunol.164:4340-4347,2000)。通过使用抗C5单克隆抗体(MoAb)还为C5和补体在RA中的重要性提供了进一步的证据。在鼠CIA模型中经腹膜内预防性给予抗C5MoAb几乎完全防止了疾病的发生,而在活动性关节炎期间的治疗则产生显著的临床益处和更轻微的组织学疾病(Wang,Y.等,Proc.Natl.Acad.Sci.USA 92:8955-59,1995)。
通过使用最近开发出的一种炎性关节炎模型K/BxN T细胞受体转基因小鼠的研究,提供了关于补体活化在疾病发病机制中的潜在作用的更深入的认识(Korganow,A.S.等,Immunity 10:451-461,1999)。所有K/BxN动物都自发产生自身免疫性疾病,该病具有人RA的大多数(尽管不是全部)临床、组织学和免疫学特征。此外,将患关节炎的K/BxN小鼠的血清转移到健康动物中,通过关节炎形成免疫球蛋白(arthritogenic immunoglobulin)的转移在几天之内便引起关节炎。为了鉴定疾病发生所需要的特定补体活化步骤,将关节炎K/BxN小鼠的血清转移到各种遗传上缺失特定补体途径产物的小鼠中(Ji,H.等,Immunity 16:157-68,2002)。有趣的是,研究结果证明替代途径活化是至关重要,而经典途径活化则可有可无。此外,C5a的形成至关重要,因为C5缺陷型小鼠和C5aR缺陷型小鼠均受到保护免于发病。与这些结果相一致的是,以前的研究曾报道了C5a受体表达的遗传缺失保护小鼠不患关节炎(Grant,E.P.等,J.Exp.Med.196:1461-1471,2002)。
美国康涅狄格州纽黑文市的Alexion Pharmaceuticals公司正在开发防止人补体成分C5裂解成其促炎成分的人源化抗C5MoAb(5G1.1),作为RA的潜在治疗方法。
两个研究小组独立地提出,凝集素途径通过MBL与特异性IgG糖形(glycoform)的相互作用促进RA患者的炎症(Malhotra等,Nat.Med.1:237-243,1995;Cuchacovich等,J.Rheumatol.23:44-51,1996)。RA与分子的Fc区缺乏半乳糖的IgG糖形(称作IgG0糖形)的显著增加有关(Rudd等,Trends Biotechnology 22:524-30,2004)。IgG0糖形的百分比随疾病进程而增加,并且当患者进入缓解期时回复到正常。在患RA的个体中,IgG0体内沉积在滑膜组织上,MBL则以高水平存在于滑液中。聚集在与RA有关的成簇IgG上的无半乳糖IgG(IgG0),可以与甘露糖结合凝集素(MBL)结合并激活补体的凝集素途径。此外,着眼于RA患者中的MBL等位变体的最新临床研究的结果表明,MBL在该病中可具有促炎作用(Garred等,J.Rheumatol.27:26-34,2000)。因此,凝集素途径在RA发病机制中可具有重要作用。
系统性红斑狼疮(SLE)是一种病因不明的自身免疫性疾病,它导致产生自身抗体、形成循环免疫复合物以及暂时不受控制地激活补体系统。尽管SLE中自身免疫性的起因仍然令人困惑,但是现在有相当多的资料表明补体活化是造成该病血管损伤的重要机制(Abramson,S.B.等,Hospital Practice 33:107-122,1998)。该病中涉及补体的经典途径和替代途径两者的活化,C4d和Bb都是中度至严重狼疮疾病活性的敏感标志(Manzi,S.等,Arthrit.Rheumat.39:1178-1188,1996)。妊娠期间替代补体途径的活化伴有系统性红斑狼疮疾病突发(Buyon,J.P.等,Arthritis Rheum.35:55-61,1992)。此外,凝集素途径可能也是引起疾病发生的原因,因为最近在SLE患者的血清中鉴定出针对MBL的自身抗体(Seelen,M.A.等,Clin Exp.Immunol.134:335-343,2003)。
认为通过经典途径的免疫复合物介导的补体活化是SLE患者中发生组织损伤的一种机制。然而,经典途径补体成分在遗传上的缺乏增加了狼疮和狼疮样疾病的风险(Pickering,M.C.等,Adv.Immunol.76:227-324,2000)。80%以上发生SLE或相关综合征的人完全缺乏C1q、C1r/C1s、C4或C3。这表示狼疮中协调补体有害效应与保护性效应时存在明显的相互矛盾。
经典途径的重要活性似乎是促进免疫复合物通过单核吞噬系统而从循环和组织中去除(Kohler,P.F.等,Am.J.Med.56:406-11,1974)。此外,最近发现在凋亡小体的去除和处理中具有重要作用的补体(Mevorarch,D.等,J.Exp.Med.188:2313-2320,1998)。经典途径功能的缺乏可能使得形成一种循环使得受试者容易发生SLE,在这种循环中,免疫复合物或凋亡细胞在组织中累积,引起炎症并释放自身抗原,继而又刺激自身抗体和更多免疫复合物的产生,从而引起自身免疫应答(Botto,M.等,Nat.Genet.19:56-59,1998;Botto,M.,ArthritisRes.3:201-10,2001)。然而,经典途径成分的这种“完全”缺乏状态只存在于大约百分之一的SLE患者中。因此,在绝大多数SLE患者中,经典途径成分的完全缺乏并不对疾病病因产生影响,补体活化可能是导致SLE发病机制的重要机制。很少有遗传上永久缺失经典途径成分的个体经常在他们生命中的某个时间发生SLE,这一事实证明冗余机制能够引起疾病。
SLE动物模型的结果支持补体活化在疾病发病机制中的重要作用。使用阻断抗C5MoAb来抑制C5活化,减轻了SLE小鼠模型NZB/NZW F1小鼠的蛋白尿和肾病(Wang Y.等,Proc.Natl.Acad.Sci.USA93:8563-8,1996)。此外,与未治疗对照相比,用抗C5MoAb治疗植入抗DNA抗体分泌细胞的患有严重联合免疫缺陷病的小鼠,导致了蛋白尿和肾组织学图像的改善,并伴有存活率相关益处(Ravirajan,C.T.等,Rheumatology 43:442-7,2004)。替代途径也在SLE的自身免疫性疾病表现上具有重要作用,因为将B因子缺陷型小鼠与SLE的MRL/lpr模型回交时,显示缺乏B因子减轻了血管炎、肾小球病,降低了常见于这种模型的C3消耗和IgG3RF水平,而不改变其它自身抗体水平(Watanabe,H.等,J.Immunol.164:786-794,2000)。作为SLE潜在治疗手段的人源化抗C5MoAb正在研究之中。这种抗体防止将C5裂解成C5a和C5b。在I期临床试验中,没有发现严重不利作用,为了确定其在SLE中的功效,更多的人体试验正在进行之中(Strand,V.,Lupus 10:216-221,2001)。
人和动物研究的结果都支持补体系统是直接引起肌营养不良发病机制的原因的可能性。人营养不良肌活检样品(dystrophic biopsies)的研究表明,C3和C9沉积在营养不良肌肉坏死和未坏死的纤维上(Cornelio和Dones,Ann.Neurol.16:694-701,1984;Spuler和Engel,A.G.,Neurology50:41-46,1998)。Porter及其同事采用DNA微阵列方法,观察到在符合营养不良疾病发生的肌养蛋白缺陷型(mdx)小鼠中,多数补体相关的mRNA的基因表达显著增加(Porter等,Hum.Mol.Genet.77:263-72,2002)。
人dysferlin(一种跨膜肌肉蛋白)编码基因的突变,已被鉴定为两种形式的骨骼肌肉疾病,即肢带肌营养不良(LGMD)和Miyoshi肌病的主要风险因素(Liu等,Nat.Genet.20:31-6,1998)。已经研发出几个具有dysferlin突变的小鼠模型,并且它们还发生进行性肌营养不良。已经在一些LGMD患者的未坏死肌肉纤维表面上鉴定出补体级联的活化(Spuler和Engel.,Neurology50:41-46,1998)。在近期的研究中,Wenzel及其同事证实,鼠和人缺乏dysferlin的肌肉纤维均缺乏补体抑制因子CD33/DAF,该抑制因子是一种特异性C5b-9MAC(膜攻击复合物)抑制剂(Wenzel等,J.Immunol.175:6219-25,2005)。因而,缺乏dysferlin的未坏死肌肉细胞更易受补体介导的细胞裂解的影响。Wenzel及其同事提出,骨骼肌肉细胞的补体介导的裂解可能是参与在患者中发生LGMD和Miyoshi肌病的主要病理机制。Connolly及其同事研究了补体C3在严重先天性营养不良模型dy-l-小鼠的发病机制中的作用,该小鼠缺乏层粘连蛋白α2(Connolly等,J.Neuroimmunol.127:80-7,2002)。他们开发出遗传上缺乏C3和层粘连蛋白α2两者的动物,并且发现缺乏C3延长了肌营养不良的dy-l-模型的存活。此外,双敲除(C3-l-,dy-l-)小鼠比dy-l-小鼠表现出更强的肌肉力量。该项研究表明,补体系统可能直接促成先天性营养不良这种形式的发病机制。
因此,本发明的一个方面涉及通过将包含治疗有效量的MASP-2抑制剂在药物载体中的组合物给予患有炎性和非炎性关节炎以及其它肌肉骨骼疾病的受试者来预防或治疗这些疾病,所述疾病包括但不限于骨关节炎、类风湿性关节炎、青少年类风湿性关节炎、痛风、神经病性关节病、银屑病性关节炎、关节强硬性脊椎炎或其它脊椎关节病以及结晶性关节病、肌营养不良或系统性红斑狼疮(SLE)。可以全身性给予受试者MASP-2抑制剂,例如通过动脉内、静脉内、肌内、皮下或其它胃肠外给药,或者对于非肽能药物可通过口服给药。或者,可通过局部递药,例如通过关节内注射。可以在一段长的时间内定期给予MASP-2抑制剂,以治疗或控制慢性病,或者可以在包括进行关节外科手术在内的急性创伤或损伤之前、期间和/或之后的一段时间单次或重复给药。
肾疾病
各种各样的肾疾病的发病机制中涉及补体系统的活化,包括肾小球系膜增生性肾小球肾炎(IgA肾病、贝惹病(Berger's disease))(Endo,M.等,Clin.Nephrology55:185-191,2001)、膜性肾小球肾炎(Kerjashki,D.,Arch B Cell Pathol.58:253-71,1990;Brenchley,P.E.等,Kidney Int.,41:933-7,1992;Salant,D.J.等,Kidney Int.35:976-84,1989)、膜增生性肾小球肾炎(肾小球系膜毛细血管性肾小球肾炎)(Bartlow,B.G.等,Kidney Int.15:294-300,1979;Meri,S.等,J.Exp.Med.175:939-50,1992)、急性感染后肾小球肾炎(链球菌感染后肾小球肾炎)、冷球蛋白血症性肾小球肾炎(Ohsawa,I.等,Clin Immunol.101:59-66,2001)、狼疮肾炎(Gatenby,P.A.,Autoimmunity 11:61-6,1991)和亨诺赫–舍恩莱因紫癜肾炎(Endo,M.等,Am.J.Kidney Dis.35:401-407,2000)。对肾疾病中所涉及的补体的认识已有几十年,但是有关它在肾疾病的发作、进展和消退期的确切作用仍存在较多争议。在正常条件下,补体的影响对宿主是有利的,但是补体的不当活化和沉积可能引起组织损伤。
有充分的证据表明,肾小球炎症即肾小球肾炎常常是由于免疫复合物沉积在肾小球或肾小管结构上引发的,继而又引发了补体活化、炎症和组织损伤。Kahn和Sinniah证实,取自各种形式肾小球肾炎患者的活检组织的肾小管基底膜上,C5b-9的沉积增加(Kahn,T.N.等,Histopath.26:351-6,1995)。在IgA肾病患者的研究中(Alexopoulos,A.等,Nephrol.Dial.Transplant 10:1166-1172,1995),C5b-9在肾小管上皮/基底膜结构上的沉积与血浆肌酸酐水平相关。另一项膜性肾病的研究证实了临床结果与尿sC5b-9水平之间的关系(Kon,S.P.等,Kidney Int.48:1953-58,1995)。sC5b-9水平升高与预后不良呈正相关。Lehto等人测得膜性肾小球肾炎患者尿中CD59以及C5b-9水平升高(Lehto,T.等,Kidney Int.47:1403-11,1995),CD59是一种抑制质膜中膜攻击复合物的补体调节因子。取自这些相同患者的活检组织样品的组织病理学分析证实,C3和C9蛋白在肾小球中沉积,而C59在这些组织中的表达比正常肾组织的表达减少。这些不同的研究表明,正在发生补体介导的肾小球肾炎导致尿中排泄出补体蛋白,这与组织损伤程度和疾病预后的程度相互关联。
抑制各种肾小球肾炎动物模型的补体活化也证实了该病病因中补体活化的重要性。在膜增生性肾小球肾炎(MPGN)模型中,C6缺陷型大鼠(不能形成C5b-9)中输注抗Thy1抗血清导致肾小球细胞增殖减少90%,血小板和巨噬细胞浸润降低80%,IV型胶原合成减少(肾小球基质扩展的标志)以及蛋白尿比C6+正常大鼠的少50%(Brandt,J.等,Kidney Int.49:335-343,1996)。这些结果表明在这种大鼠抗胸腺细胞血清模型中,C5b-9是补体导致的组织损伤的主要介质。在另一种肾小球肾炎模型中,输注分级剂量的兔抗大鼠肾小球基底膜,产生了多形核白细胞(PMN)剂量依赖性地流入,这种多形核白细胞流入通过预先用眼镜蛇毒因子处理(消耗补体)而被减弱(Scandrett,A.L.等,Am.J.Physiol.268:F256-F265,1995)。眼镜蛇毒因子处理的大鼠也显示出组织病理减轻、长期蛋白尿减少以及肌酸酐水平比对照大鼠的降低。Couser等人采用三种GN大鼠模型(抗胸腺细胞血清、Con A抗Con A和被动性海曼肾炎(passive Heymann nephritis)),证实使用重组sCR1蛋白抑制补体这类方法的潜在治疗功效(Couse,W.G.等,J.Am.Soc.Nephrol.5:1888-94,1995)。比起对照大鼠,用sCR1处理的大鼠则出现PMN、血小板和巨噬细胞流入显著减少、肾小球系膜裂解(mesangiolysis)和蛋白尿减少。通过在NZB/W F1小鼠模型中使用抗C5MoAb,为补体活化在肾小球肾炎中的重要性提供了进一步的证据。抗C5MoAb抑制C5的裂解,从而阻断C5a和C5b-9的产生。用抗C5MoAb连续治疗6个月,导致肾小球肾炎进程显著改善。美国康涅狄格州纽黑文市的Alexion Pharmaceuticals公司正在开发一种防止人补体成分C5裂解成其促炎成分的人源化抗C5MoAb单克隆抗体(5G1.1),作为肾小球肾炎潜在的治疗方法。
通过对遗传上缺失特定补体成分的患者的研究,为补体在肾脏损伤中的病理学作用提供了直接证据。许多报道证明了肾疾病与补体调节因子H缺乏有关(Ault,B.H.,Nephrol.14:1045-1053,2000;Levy,M.等,Kidney Int.30:949-56,1986;Pickering,M.C.等,Nat.Genet.31:424-8,2002)。H因子的缺乏导致B因子和C3的低血浆水平以及C5b-9的消耗。非典型性膜增生性肾小球肾炎(MPGN)和特发性溶血性尿毒症综合征(HUS)都与H因子的缺乏有关。H因子缺陷型猪(Jansen,J.H.等,Kidney Int.53:331-49,1998)和H因子敲除小鼠(Pickering,M.C.,2002)都表现出MPGN样症状,这就证实了H因子在补体调节中的重要性。其它补体成分的缺乏与系统性红斑狼疮(SLE)发生后的继发性肾病有关(Walport,M.J.,Davies等,Ann.N.Y.Acad.Sci.815:267-81,1997)。C1q、C4和C2的缺乏使得非常容易通过与缺乏清除免疫复合物和凋亡物质有关的机制而发生SLE。在这些SLE患者中许多发生了狼疮肾炎,其特征在于免疫复合物沉积在整个肾小球中。
通过鉴定患者体内针对补体成分的自身抗体(其中一些与肾疾病直接相关),为补体活化与肾疾病关联提供了进一步的证据(Trouw,L.A.等,Mol.Immunol.38:199-206,2001)。这些自身抗体的许多种与肾疾病的关联性如此高,以致于引入了术语肾炎因子(NeF)来表示这种活性。在临床研究中,肾炎因子阳性患者中大约有50%发生MPGN(Spitze,R.E.等,Clin.Immunol.Immunopathol.64:177-83,1992)。C3NeF是针对替代途径C3转化酶(C3bBb)的自身抗体,它使这种转化酶稳定,从而促进替代途径活化(Daha,M.R.等,J.Immunol.116:1-7,1976)。同样地,对经典途径C3转化酶(C4b2a)具有特异性的自身抗体(称为C4NeF)使该转化酶稳定,从而促进经典途径活化(Daha,M.R.等,J.Immunol.125:2051-2054,1980;Halbwachs,L.等,J.Clin.Invest.65:1249-56,1980)。已表明,抗C1q自身抗体与SLE患者的肾炎有关(Hovath,L.等,Clin.Exp.Rheumatol.19:667-72,2001;Siegert,C.等,J.Rheumatol.18:230-34,1991;Siegert,C.等,Clin.Exp.Rheumatol.10:19-23,1992),还有报道指出这些抗C1q自身抗体的效价升高被用来预测肾炎的突发(Coremans,I.E.等,Am.J.Kidney Dis.26:595-601,1995)。从SLE患者尸检肾洗脱出的免疫沉积物揭示这些抗C1q自身抗体的累积(Mannick,M.等,Arthritis Rheumatol.40:1504-11,1997)。所有这些事实都指向这些自身抗体的病理学作用。然而,并不是所有具有抗C1q自身抗体的患者都发生肾病,一些健康个体中也有低效价的抗C1q自身抗体(Siegert,C.E.等,Clin.Immunol.Immunopathol.67:204-9,1993)。
除了补体活化的替代途径和经典途径以外,凝集素途径在肾疾病中也有重要的病理学作用。通过免疫组织化学技术,从获自诊断为患有几种不同肾病的患者的肾活检材料中,检测出高水平的MBL、MBL相关的丝氨酸蛋白酶及补体活化产物,这些肾病包括亨诺赫–舍恩莱因紫癜肾炎(Endo,M.等,Am.J.Kidney Dis.35:401-407,2000)、冷球蛋白血症性肾小球肾炎(Ohsawa,I.等,Clin.Immunol.101:59-66,2001)以及IgA肾病(Endo,M.等,Clin.Nephrology 55:185-191,2001)。因此,尽管已经清楚补体与肾病之间的相关性这一事实几十年,但是关于补体如何确切影响这些肾病的数据仍远远不够完整。
因此,本发明的一个方面涉及通过将包含治疗有效量的MASP-2抑制剂在药物载体中的组合物,给予患有肾病的受试者以治疗这类疾病,所述疾病包括但不限于肾小球系膜增生性肾小球肾炎、膜性肾小球肾炎、膜增生性肾小球肾炎(肾小球系膜毛细血管性肾小球肾炎)、急性感染后肾小球肾炎(链球菌感染后肾小球肾炎)、冷球蛋白血症性肾小球肾炎、狼疮肾炎、亨诺赫–舍恩莱因紫癜肾炎或者IgA肾病。可以全身性给予受试者MASP-2抑制剂,例如通过动脉内、静脉内、肌内、皮下或其它胃肠外给药,或者对于非肽能药物可通过口服给药。可以在一段长的时间内定期给予MASP-2抑制剂,以治疗或控制慢性病,或者可以在急性创伤或损伤之前、期间或之后的一段时间单次或重复给药。
皮肤疾病
银屑病是慢性虚弱性皮肤病,累及数百万人,是遗传和环境因素两者的结果。一般认为局部用药以及UVB和PUVA光线疗法是银屑病的一线治疗方法。然而,对于遍及全身的或更大范围的疾病来说,全身性治疗指示为初步治疗,或者在一些情况下用以增强UVB和PUVA治疗。
各种皮肤疾病(例如银屑病)的潜在病因支持免疫和促炎过程(包括补体系统的参与)的作用。此外,已经确定了补体系统作为重要的非特异性皮肤防御机制的作用。它的活化导致不仅有助于维持正常的宿主防御、而且还介导炎症和组织损伤的产物的产生。补体的促炎产物包括具有调理活性和细胞刺激活性的C3大片段(C3b和C3bi)、低分子量的过敏毒素(C3a、C4a和C5a)和膜攻击复合物。其中,C5a或其降解产物脱Arg的C5a似乎是最重要的介质,因为它发挥对炎性细胞的有效趋化效应。真皮内给予C5a过敏毒素诱导的皮肤变化与通过免疫复合物介导的补体活化而发生的皮肤超敏血管炎中所观察到的变化十分相似。补体活化参与了自身免疫性大疱性皮肤病中炎症变化的发病机制。由表皮天疱疮抗体引起的补体活化似乎是称为嗜酸细胞性海绵样水肿这一特征性炎症变化发生的原因。在大疱性类天疱疮(BP)中,基底膜区抗原与BP抗体之间的相互作用导致了补体活化,似乎与嵌入真皮表皮接界的白细胞有关。所产生的过敏毒素不仅激活浸润性白细胞,而且还诱导肥大细胞脱粒,这就促使真皮表皮分离和嗜酸性粒细胞浸润。同样地,补体活化似乎在获得性大疱性表皮松解和妊娠疱疹中所观察到的真皮表皮分离中发挥更直接的作用。
补体参与银屑病的证据来自于文献中记载的涉及银屑病和相关疾病的炎症变化的病理生理学机制的最新实验发现。越来越多的证据表明,T细胞介导的免疫力在银屑病病变的引发和维持中起着重要作用。有研究显示,银屑病病变中由活化T细胞产生的淋巴因子对表皮增殖具有强大的影响力。在银屑病病变的严重发炎部位能够观察到角质层下的特征性嗜中性粒细胞累积。通过由受刺激的角质形成细胞释放的趋化因子、IL-8和Gro-α的协同作用,特别是通过替代补体途径活化所产生的C5a/脱arg的C5a,化学趋化性地吸引并激活嗜中性粒细胞(Terui,T.,TahokuJ.Exp.Med.190:239-248,2000;Terui,T.,Exp.Dermatol.9:1-10,2000)。
银屑病鳞屑提取物含有独特的趋化肽部分,它可能参与诱导节律性经表皮的白细胞趋化反应。最新研究鉴定出该部分中存在两种不相关的趋化肽,即C5a/脱Arg的C5a和白介素8(IL-8)及其相关细胞因子。为研究它们对经表皮白细胞迁移的相对影响以及它们在银屑病病变中的相互关系,对银屑病病变鳞屑提取物和相关的无菌脓疱皮肤病提取物中的免疫反应性C5a/脱Arg的C5a和IL-8浓度进行了定量。发现病变皮肤角质组织提取物中的C5a/脱Arg的C5a和IL-8的浓度比非炎性正常角质皮肤(orthokeratotic skin)的浓度显著增加。C5a/脱Arg的C5a浓度的增加是病变鳞屑提取物特有的。根据这些结果,似乎C5a/脱Arg的C5a仅在优先利于补体活化的特定情况下在炎性病变皮肤中产生。这就为使用补体活化抑制剂改善银屑病病变提供了理论基础。
虽然有研究显示补体系统的经典途径在银屑病中被激活,但是仍有少量关于替代途径参与银屑病炎症反应的报道。在补体活化途径的传统观点范围内,补体片段C4d和Bb分别在经典途径和替代途径活化时被释放。因此,C4d或Bb片段的存在表示通过经典途径和/或替代途径进行的补体活化。一项研究使用酶免疫测定技术测定了银屑病鳞屑提取物中C4d和Bb的水平。这些皮肤病鳞屑含有的用酶免疫测定法可检测到的C4d和Bb水平比非炎性皮肤角质层中的高(Takematsu,H.等,Dermatologica 181:289-292,1990)。这些结果表明在银屑病的病变皮肤中除补体的经典途径之外,替代途径也被激活。
通过对患有特应性皮炎(AD)的35名患者和患有轻微至中度银屑病的24名患者的外周血中的正常补体成分和活化产物进行测定,获得补体参与银屑病和特应性皮炎的额外证据。血清中C3、C4和C1钝化物(C1INA)的水平用放射免疫扩散测定,而C3a和C5a水平则用放射免疫测定法测定。观察到与健康的非特应性对照相比,两种疾病中的C3、C4和C1INA的水平都显著增加。在AD中,C3a水平趋于升高,而在银屑病中,C3a水平则显著提高。这些结果表明,在AD和银屑病中,补体系统都参与了炎症过程(Ohkonohchi,K.等,Dermatologica 179:30-34,1989)。
银屑病的病变皮肤中补体活化还导致末端补体复合物在表皮内沉积,如通过测定银屑病患者的血浆和角质组织中的SC5b-9水平所显示的一样。已经发现银屑病血浆中的SC5b-9水平显著高于对照或者特应性皮炎患者的水平。得自病变皮肤的总蛋白提取物的研究显示,尽管在非炎性角质组织中无法检测到SC5b-9,但在银屑病的病变角质组织中有高水平的SC5b-9。通过使用抗C5b-9新抗原的单克隆抗体的免疫荧光法,仅在银屑病皮肤的角质层中观察到C5b-9的沉积。总的来说,在银屑病的病变皮肤中,补体系统被激活,补体活化一直进行到终端步骤,产生了膜攻击复合物。
近来,新的选择性靶向免疫系统的生物药物已经可用来治疗银屑病。目前已被美国FDA批准或者处于III期研究的四种生物药物是:阿来西普(alefacept)(艾默)和efalizuMoAb(依法珠单抗)(它们是T细胞调节剂);依那西普(etanercept)(恩利)(它是可溶性TNF受体);以及inflixiMoAb(它是抗TNF单克隆抗体)。依法珠单抗(Raptiva)是免疫应答调节剂,其中作用的靶向机制是阻断淋巴细胞上的LFA-1与抗原呈递细胞和血管内皮细胞上的ICAM-1之间的相互作用。通过依法珠单抗(Raptiva)结合CD11a使淋巴细胞上可用的CD11a结合位点饱和,使淋巴细胞上细胞表面CD11a的表达下调。这种作用机制抑制了T细胞活化、细胞至真皮和表皮的迁移以及T细胞再活化。因此,大量科学证据表明了补体在皮肤的炎性疾病状态中的作用,最新制药方法现已针对免疫系统或特定的炎症过程。然而,还没有研究将MASP-2视作靶向目标。根据本发明的发明人对MASP-2在补体活化中的作用的新认识,本发明的发明人认为MASP-2是治疗银屑病和其它皮肤病的有效靶标。
因此,本发明的一个方面涉及治疗银屑病、自身免疫性大疱性皮肤病、嗜酸细胞性海绵样水肿、大疱性类天疱疮、获得性大疱性表皮松解、特应性皮炎、妊娠疱疹和其它皮肤病,以及用于治疗热烧伤和化学烧伤,包括由此引起的毛细血管渗漏,即通过将包含治疗有效量的MASP-2抑制剂在药物载体中的组合物给予患有所述皮肤病的受试者进行治疗。MASP-2抑制剂可以通过应用含有MASP-2抑制剂的喷雾剂、洗剂、凝胶剂、糊剂、软膏剂或灌洗溶液剂,局部给予受试者,或者例如通过动脉内、静脉内、肌内、皮下或其它胃肠外给药等全身性给予,或者对于非肽能药物可通过口服给药。对于急性病,治疗可包括单次给药或重复用药或给药,或者对于控制慢性病,则包括定期用药或给药。
移植
补体系统的活化显著地促进了实体器官移植后的炎症反应。在同种异体移植中,补体系统可被缺血/再灌注激活,并且可能被针对移植物的抗体激活(Baldwin,W.M.等,Springer Seminol Immunopathol.25:181-197,2003)。从非灵长类至灵长类的异种移植中,补体的主要激活因子是预先存在的抗体。动物模型中的研究表明使用补体抑制剂可显著延长移植物的存活(参见下文)。因此,补体系统在器官移植后的器官损伤中有着明确的作用,从而本发明的发明人认为使用针对MASP-2的补体抑制剂可预防同种异体移植或异种移植后对移植物的损伤。
先天免疫机制、特别是补体在针对移植物的炎症反应和免疫应答中所起的作用比以前所认识的作用要大。例如,替代补体途径活化似乎介导了肾缺血/再灌注损伤,近端小管细胞可能是这种环境中补体成分的来源兼攻击部位。肾脏内局部产生的补体也在针对移植物的细胞和抗体介导的免疫应答两者的发生中都发挥着作用。
C4d是活化补体因子C4的降解产物,C4是经典途径和凝集素依赖途径的成分。C4d染色已经成为急性病和慢性病环境中体液排斥的有用标记,重新引起对抗供体抗体形成的重要性的关注。C4d和急性细胞排斥的形态学标志之间的关联性具有统计显著性。在24-43%的I型发作、45%的II型排斥和50%的III型排斥中观察到C4d(Nickeleit,V.等,J.Am.Soc.Nephrol.13:242-251,2002;Nickeleit,V.等,Nephrol.Dial.Transplant 18:2232-2239,2003)。正在开发抑制补体或降低局部合成作为移植后达到改善临床结果的手段的多种治疗方法。
补体级联活化是由于移植期间的许多过程而发生的。目前的疗法尽管在限制细胞排斥方面是有效的,但是并不能完全应付所面临的所有障碍。这些障碍包括体液排斥和慢性同种异体移植肾病或功能障碍。尽管对移植器官的总体反应是宿主方许多效应机制的结果,不过补体可能在其中一些方面起着关键作用。在肾移植环境下,近端小管细胞的局部补体合成似乎特别重要。
补体特异性抑制剂的可获得性可能为改善器官移植后的临床结果提供了机会。通过阻滞补体攻击的机制起作用的抑制剂可能是特别有用的,因为它们有望提高功效,并且避免无免疫应答接受者的系统性补体耗尽。
补体在异种移植排斥中也起着关键作用。因此,有效的补体抑制剂作为潜在的治疗药是非常令人关注的。在猪-灵长类器官移植中,超急性排斥(HAR)是由抗体沉积和补体活化引起的。已经对预防猪-灵长类组合中超急性异种移植物排斥的多项策略和目标进行了试验。这些方法通过去除天然抗体、用眼镜蛇毒因子耗尽补体、或者用可溶性补体抑制剂sCR1防止C3活化得以实现。此外,补体活化阻断剂(CAB-2),一种源自人衰变加速因子(DAF)和膜辅因子蛋白的重组可溶性嵌合蛋白,抑制经典途径和替代途径两者的C3和C5转化酶。CAB-2减少用人血离体灌注猪心的补体介导的组织损伤。将猪心异位移植到未接受任何免疫抑制的猕猴中时对CAB-2功效的研究表明,在接受CAB-2的猴中,移植物存活期显著延长(Salerno,C.T.等,Xenotransplantation 9:125-134,2002)。CAB-2显著抑制补体活化,体现在C3a和SC5b-9的产生急剧减少。在移植排斥时,iC3b、C4和C9的组织沉积与对照的沉积类似或者略微有减少,IgG、IgM、C1q和纤维蛋白沉积没有变化。因此,这种补体抑制方法消除了移植到猕猴体内的猪心的超急性排斥。这些研究证实了补体抑制对存活的有益效果,本发明的发明人认为MASP-2抑制剂还可用于异种移植。
另一种方法集中在确定抗补体5(C5)单克隆抗体能否在大鼠-预敏化小鼠心脏移植模型中防止超急性排斥(HAR),以及这些MoAb与环孢菌素(cyclosporine)和环磷酰胺联用能否实现移植物的长期存活。研究表明抗C5MoAb预防HAR(Wang,H.等,Transplantation68:1643-1651,1999)。因此本发明的发明人认为,补体级联中的其它靶(例如MASP-2)对于在未来的临床异种移植中防止HAR和急性血管排斥也可能是有价值的。
虽然补体在见于异种移植的超急性排斥中的关键作用已经非常确定,但是在同种异体移植中的不太明显的作用开始显现。早已清楚补体和获得性免疫应答之间的联系,其中发现缺乏补体的动物在抗原刺激后产生低于正常的抗体应答。研究表明抗原用补体裂解产物C3d的调理作用极大地增加了抗原呈递到B细胞的有效性,并且表明是通过2型补体受体连接到某些B细胞上而起作用的。这项工作已经延伸到小鼠皮肤移植模型中的移植环境,其中C3和C4缺陷型小鼠在同种抗体的产生方面具有明显的缺点,这是由于类别转换成高亲和性IgG失败所致。由于抗供体抗体和体液排斥的重要性,因此这些机制在肾移植中的重要性随之增加。
以前的工作已证实,在肾脏移植之后的同种异体移植物排斥期间,近端小管细胞的C3合成上调。在小鼠肾脏移植模型中,已对局部合成补体的作用进行了研究。与C3阳性供体的对照移植物相比,将C3阴性供体的移植物移植到C3充足的受体中,显示其存活期延长(>100天),而对照移植物14天内就被排斥。此外,与对照的相比,C3阴性移植物的接受者体内抗供体T细胞增值应答显著降低,这表明局部合成的C3对T细胞初敏的作用。
这些观察结果表明这样的可能性,即供体抗原暴露于T细胞首先是在移植物中发生的,而且局部合成的补体通过供体抗原的调理作用或者通过向抗原呈递细胞或T细胞提供另外的信号而增强了抗原呈递。在肾脏移植环境下,产生补体的肾小管细胞也表明补体沉积在它们的细胞表面上。
因此,本发明的一个方面涉及通过将包含治疗有效量的MASP-2抑制剂在药物载体中的组合物给予移植受体,来预防或治疗由组织或实体器官移植引起的炎症反应,所述移植受体包括已接受完整器官(例如肾、心、肝、胰腺、肺、角膜等)或移植物(如瓣膜、腱、骨髓等)的同种异体移植或异种移植的受试者。可以通过动脉内、静脉内、肌内、皮下或其它胃肠外给药给予受试者MASP-2抑制剂,或者对于非肽能抑制剂可口服给予。给药可发生在移植后的急性期和/或作为长期移植后疗法。附加于移植后给药或者代替移植后给药,可在移植之前和/或移植手术期间用MASP-2抑制剂治疗受试者,和/或用MASP-2抑制剂预处理待移植的器官或组织。器官或组织的预处理可能需要将含有MASP-2抑制剂的溶液剂、凝胶剂或糊剂经喷雾或冲洗表面而应用到器官或组织表面,或者可将器官或组织浸泡在含有MASP-2抑制剂的溶液中。
中枢与外周神经系统疾病和损伤
各种中枢神经系统(CNS)或外周神经系统(PNS)疾病或损伤的发病机制涉及补体系统的活化,所述疾病或损伤包括但不限于多发性硬化(MS)、重症肌无力(MG)、亨廷顿病(HD)、肌萎缩性侧索硬化(ALS)、急性热病性多神经炎、中风后再灌注、椎间盘退行性疾病、脑外伤、帕金森病(PD)和阿尔茨海默病(AD)。补体蛋白在包括神经元、星形胶质细胞和小胶质细胞在内的CNS细胞中合成的初步测定,以及对补体活化之后在CNS中产生的过敏毒素能改变神经元功能的认识,揭示了补体在CNS疾病中的潜在作用(Morgan,B.P.等,Immunology Today17:10:461-466,1996)。现已表明,C3a受体和C5a受体存在于神经元上,并广泛分布在感觉、运动和边缘脑系统的各个不同部分上(Barum,S.R.,Immunologic Research 26:7-13,2002)。而且,已表明,过敏毒素C5a和C3a改变啮齿动物的饮食习惯,并能诱导小胶质细胞和神经元的钙信号转导。这些发现增加了有关在各种CNS炎性疾病中抑制补体活化的治疗效用的可能性,所述疾病包括脑外伤、脱髓鞘、脑膜炎、中风和阿尔茨海默病。
脑外伤或脑出血是常见的临床问题,补体活化可能产生并加重所形成的炎症和水肿。已在大鼠脑外伤模型中对补体抑制作用进行了研究(Kaczorowski等,J.Cereb.Blood Flow Metab.15:860-864,1995)。在临脑损伤之前给予sCR1显著抑制嗜中性粒细胞浸润到受伤区域,这表明了补体对于吞噬细胞募集的重要性。同样地,血浆和脑脊液(CSF)中高水平的多种补体活化产物的存在清楚表明了脑出血之后患者中的补体活化。补体活化和C5b-9复合物染色增加在隔离的腰椎间盘组织中得到证实,可能表明了在椎间盘突出组织诱发的坐骨神经痛中的作用(Gronblad,M.等,Spine 28(2):114-118,2003)。
MS(多发性硬化)的特征在于CNS内将轴突包鞘并绝缘的髓磷脂逐渐丧失。尽管尚不清楚最初的原因,不过有大量证据表明涉及到免疫系统(Prineas,J.W.等,Lab Invest.38:409-421,1978;Ryberg,B.,J.Neurol.Sci.54:239-261,1982)。还有明确的证据表明,补体在CNS或PNS脱髓鞘疾病的病理生理学中起着突出作用,这些疾病包括MS、急性热病性多神经炎和米勒-费希尔综合征(Gasque,P.等,Immunopharmacology 49:171-186,2000;Barnum,S.R.in Bondy S.等(主编)Inflammatory events in neurodegeneration,Prominent Press,pp.139-156,2001)。补体是引起组织破坏、发炎、髓磷脂碎片的清除甚至是轴突髓鞘再形成的原因。尽管补体参与证据确凿,但是补体治疗性靶的鉴定目前仍只是在实验性变应性脑脊髓炎(EAE)(一种多发性硬化的动物模型)中进行评价。研究表明,与EAE对照小鼠相比,缺乏C3或B因子的EAE小鼠脱髓鞘作用减弱(Barnum,Immunologic Research 26:7-13,2002)。使用称为“sCrry”的可溶形式的补体抑制剂和C3-/-以及B因子-/-的EAE小鼠研究证实补体是疾病模型在若干水平上发生和发展的原因。此外,B因子-/-小鼠中的EAE严重性的显著降低,这为补体替代途径在EAE中的作用提供了进一步的证据(Nataf等,J.Immunology165:5867-5873,2000)。
MG(重症肌无力)是乙酰胆碱受体丧失和运动终板破坏的神经肌肉接头疾病。sCR1在MG动物模型中非常有效,这进一步表明了补体在该疾病中的作用(Piddelesden等,J.Neuroimmunol.1997)。
神经退行性疾病AD(阿尔茨海默病)的组织学标志是老年斑和神经纤维原缠结(McGeer等,Res.Immunol.143:621-630,1992)。这些病理学标记对于补体系统成分也是强染色的。证据指向导致神经元死亡和认知功能障碍的局部神经炎症状态。老年斑含有异常的淀粉样β肽(Aβ,这是一种源自淀粉样前体蛋白的肽。研究表明Aβ结合C1并能引发补体活化(Rogers等,Res.Immunol.143:624-630,1992)。此外,AD的显著特征是与经典补体途径从C1q到C5b-9的活化蛋白相关,观察到它们高度定位于神经炎斑中(Shen,Y.等,Brain Research 769:391-395,1997;Shen,Y.等,Neurosci.Letters305(3):165-168,2001)。因此,Aβ不但启动了经典途径,而且所产生的连续炎症状态可能是神经元细胞死亡的原因。而且,AD中补体活化已经进行到末端C5b-9阶段的这一事实表明,补体系统的调节机制仍不能终止补体活化过程。
有研究提出了几种补体途径抑制剂作为潜在的AD治疗手段,包括作为C1Q结合抑制剂的蛋白聚糖、作为C3转化酶抑制剂和C5活化阻断剂或C5a受体抑制剂的萘莫司他(Nafamstat)(Shen,Y.等,Progress in Neurobiology,70:463-472,2003)。MASP-2作为先天补体途径起始步骤以及用于替代途径活化的作用,提供了可能的新的治疗手段,这得到了表明补体途径参与AD的大量数据的支持。
同其它CNS退行性疾病一样,在PD(帕金森病)患者脑的受损区域存在炎症的证据,所述炎症的特征在于神经胶质反应(特别是小胶质细胞反应),以及HLA-DR抗原、细胞因子和补体成分的表达增加。这些观察结果表明,免疫系统机制参与了PD中神经元损伤的发病机制。然而,还没有阐明PD中的原发性损伤的细胞机制,但是,有可能是线粒体突变、氧化应激和细胞凋亡在起作用。此外,PD中由纹状体和黑质的神经元损伤引起的炎症可能加重病程。这些观察结果表明,用补体抑制药治疗可能起着减缓PD进程的作用(Czlonkowska,A.等,Med.Sci.Monit.8:165-177,2002)。
因此,本发明的一个方面涉及通过应用包含治疗有效量的MASP-2抑制剂在药物载体中的组合物来治疗患有外周神经系统(PNS)疾病或损伤和/或中枢神经系统(CNS)疾病或损伤的受试者,来治疗这些疾病或损伤。认为可根据本发明治疗的CNS和PNS疾病和损伤包括但不限于多发性硬化(MS)、重症肌无力(MG)、亨廷顿病(HD)、肌萎缩性侧索硬化(ALS)、急性热病性多神经炎、中风后再灌注、椎间盘退行性疾病、脑外伤、帕金森病(PD)、阿尔茨海默病(AD)、米勒-费希尔综合征、脑外伤和/或出血、脱髓鞘,还可为脑膜炎。
对于治疗CNS疾病和脑外伤来说,可以通过鞘内、颅内、脑室内、动脉内、静脉内、肌内、皮下或其它胃肠外给药给予受试者MASP-2抑制剂,对于非肽能抑制剂可口服给予。可通过全身给药途径或者通过局部给予功能障碍或创伤部位来治疗PNS疾病和脑外伤。可按照医师的规定定期重复给予本发明的MASP-2抑制剂组合物,直到症状得到有效缓解或控制。
血液病
脓毒症是由于患者对侵入微生物的过度反应而引起的。补体系统的主要功能是配合对侵入细菌和其它病原体的炎症反应。与这种生理作用相一致,许多研究显示补体活化在脓毒症的发病机制中具有主要作用(Bone,R.C.,Annals.Internal.Med.115:457-469,1991)。脓毒症临床表现的定义在不断发展中。通常将脓毒症定义为对感染的系统性宿主应答。然而,在许多情况下,有脓毒症症状的患者中没有发现感染的临床证据(如血液培养细菌阳性)。这种矛盾之处最早是在1992年的共识会议上确定术语“全身炎症反应综合征”(systemic inflammatory response syndrome,SIRS)时被注意到的,SIRS不需要确定的细菌感染的存在(Bone,R.C.等,Crit.Care Med.20:724-726,1992)。目前普遍认为,伴随脓毒症和SIRS的是不能调控炎症反应。出于该简述的目的,我们会考虑脓毒症的临床定义还包括严重脓毒症、败血症性休克和SIRS。
在1980年代末期之前,脓毒症患者的主要感染源是革兰氏阴性菌。已知革兰氏阴性菌细胞壁的主要成分脂多糖(LPS)刺激炎症介质从各种细胞类型中释放出来,并且当注射到动物体内时诱导急性感染症状(Haeney,M.R.等,Antimicrobial Chemotherapy 41(增刊A):41-6,1998)。有趣的是,引起感染的微生物的范围似乎从1970年代末期和1980年代主要是革兰氏阴性菌变成了现今的主要是革兰氏阳性菌,其原因目前不清楚(Martin,G.S.等,N.Eng.J.Med.348:1546-54,2003)。
许多研究表明补体活化在介导炎症和引起休克、特别是败血症性休克和出血性休克特征中的重要性。革兰氏阴性和革兰氏阳性生物一般都促使败血症性休克。LPS是主要经由替代途径的有效的补体活化因子,尽管抗体介导的经典途径活化也有发生(Fearon,D.T.等,N.Engl.J.Med.292:937-400,1975)。革兰氏阳性细胞壁的主要成分是肽聚糖和脂磷壁酸,两种成分都是替代补体途径的有效激活因子,尽管在特异性抗体存在时,它们也能激活经典补体途径(Joine,K.A.等,Ann.Rev.Immunol.2:461-2,1984)。
脓毒症发病机制中最初涉及补体系统是当研究人员注意到过敏毒素C3a和C5a介导各种也可能发生在脓毒症期间的炎症反应时发现的。这些过敏毒素引起血管扩张和微血管通透性增加,这是在败血症性休克中起着重要作用的事件(Schumache,W.A.等,Agents Actions34:345-349,1991)。此外,过敏毒素诱导支气管痉挛、组胺从肥大细胞中释放以及血小板聚集。而且,它们对粒细胞发挥了许多作用,例如趋化、聚集、粘附、溶酶体酶的释放、毒性超氧阴离子的产生和白三烯的形成(Shin,H.S.等,Science 162:361-363,1968;Vogt,W.,Complement 3:177-86,1986)。认为这些生物学作用在脓毒症并发症例如休克或急性呼吸窘迫综合征(ARDS)的发生中起着作用(Hammerschmidt,D.E.等,Lancet 1:947-949,1980;Slotman,G.T.等,Surgery 99:744-50,1986)。此外,过敏毒素C3a水平的升高与脓毒症的致命结果有关(Hack,C.E.等,Am.J.Med.86:20-26,1989)。在一些休克动物模型中,某些补体缺陷型品系(如C5缺陷型品系)对LPS的输注效应具有更高的抗性(Hseuh,W.等,Immunol.70:309-14,1990)。
研究显示,在啮齿动物脓毒症发作期间,用抗体阻断C5a产生极大地提高了存活率(Czermak,B.J.等,Nat.Med.5:788-792,1999)。当用抗体或小分子抑制剂阻断C5a受体(C5aR)时,得到了类似的结果(Huber-Lang,M.S.等,FASEB J.16:1567-74,2002;Riedemann,N.C.等,J.Clin.Invest.110:101-8,2002)。较早期对猴子的实验研究表明,C5a的抗体阻断减少了大肠杆菌(E.coli)诱发的败血症性休克和成人呼吸窘迫综合征(Hangen,D.H.等,J.Surg.Res.46:195-9,1989;Stevens,J.H.等,J.Clin.Invest.77:1812-16,1986)。与脓毒症不太严重的患者和幸存者相比,C5a在脓毒症病人体内升高,并且与显著降低存活率和多器官衰竭有关(Nakae,H.等,Res.Commun.Chem.Pathol.Pharmacol.84:189-95,1994;Nakae等,Surg.Today 26:225-29,1996;Bengtson,A.等,Arch.Surg.123:645-649,1988)。C5a在脓毒症期间发挥其有害作用的机制还有待更详细的研究,但是最近的数据表明,脓毒症期间C5a的产生显著损害血液嗜中性粒细胞的先天免疫功能(Huber-Lang,M.S.等,J.Immunol.169:3223-31,2002)、它们引起呼吸爆发的能力及其产生细胞因子的能力(Riedemann,N.C.等,Immunity19:193-202,2003)。此外,脓毒症期间的C5a产生似乎具有促凝血效应(Laudes,I.J.等,Am.J.Pathol.160:1867-75,2002)。在脓毒症和ARDS动物模型中,补体调控蛋白CI INH也显示出功效(Dickneite,G.,Behring Ins.Mitt.93:299-305,1993)。
凝集素途径在脓毒症的发病机制中也可能有作用。已显示MBL结合临床上重要的各种微生物,包括革兰氏阴性菌和革兰氏阳性菌,并激活凝集素途径(Neth,O.等,Infect.Immun.68:688,2000)。脂磷壁酸(LTA)越来越被视作LPS的革兰氏阳性对应物(Gram-positive counterpart)。它是诱导细胞因子从单核吞噬细胞和全血中释放的有效免疫刺激剂(Morath,S.等,J.Exp.Med.195:1635,2002;Morath,S.等,Infect.Immun.70:938,2002)。最近证实,L-纤维胶凝蛋白与从许多革兰氏阳性菌(包括金黄色葡萄球菌(Staphylococcus aureus))分离的LTA特异性结合,并且激活凝集素途径(Lynch,N.J.等,J.Immunol.172:1198-02,2004)。还显示,MBL能结合得自肠球菌(Enterococcus spp)的LTA,其中多聚甘油磷酸链被糖基取代,但是不结合来自其它9种菌种(包括金黄色葡萄球菌)的LTA(Polotsky,V.Y.等,Infect.Immun.64:380,1996)。
因此,本发明一方面提供用于治疗脓毒症或由脓毒症引起的疾病的方法,该方法通过将包含治疗有效量的MASP-2抑制剂在药物载体中的组合物给予患有脓毒症或由脓毒症引起的疾病的受试者实施,所述疾病包括但不限于严重脓毒症、败血症性休克、脓毒症引起的急性呼吸窘迫综合征和全身性炎症反应综合征。本发明提供治疗其它血液病的有关方法,该方法通过将包含治疗有效量的MASP-2抑制剂在药物载体中的组合物给予患其它血液病的受试者实施,所述其它血液病包括出血性休克、溶血性贫血、自身免疫性血栓形成性血小板减少性紫癜(TTP)、溶血性尿毒症综合征(HUS)或其它骨髓/血液破坏性疾病。可以全身性给予受试者MASP-2抑制剂,例如通过动脉内、静脉内、肌内、吸入(特别是在ARDS的情况下)、皮下或其它胃肠外给药,或者对于非肽能抑制剂可口服给予。MASP-2抑制剂组合物可与一种或多种另外的治疗药联用以对抗脓毒症和/或休克的后遗症。对于晚期脓毒症或休克或者由此引起的疾病状况来说,MASP-2抑制剂组合物可适于以速效剂型给药,例如通过静脉内或动脉内推注含有MASP-2抑制剂组合物的溶液剂来给药。可按照医师规定重复给药,直到疾病消退。
泌尿生殖系统疾病
研究表明补体系统参与了几种截然不同的泌尿生殖系统疾病,包括疼痛性膀胱病、感觉性膀胱病、慢性非细菌性膀胱炎和间质性膀胱炎(Holm-Bentzen,M.等,J.Urol.138:503-507,1987)、不育(Cruz等,Biol.Reprod.54:1217-1228,1996)、妊娠(Xu,C.等,Science 287:498-507,2000)、胎儿母体耐受(Xu,C.等,Science 287:498-507,2000)和先兆子痫(Haege,M.,Int.J.Gynecol.Obstet.43:113-127,1993)。
疼痛性膀胱病、感觉性膀胱病、慢性非细菌性膀胱炎和间质性膀胱炎是未知病因和发病机制所知甚少的疾病,因此没有任何合理的治疗方法。有关膀胱的上皮和/或粘膜表层膜缺陷的致病理论,以及有关免疫紊乱的理论成为主流(Holm-Bentzen,M.等,J.Urol.138:503-507,1987)。据报道已经测试了间质性膀胱炎患者的免疫球蛋白(IgA、IgG、IgM)、补体成分(C1q、C3、C4)和C1酯酶抑制剂。补体成分C4的血清水平有非常显著的降低(p<0.001),免疫球蛋白G显著增加(p<0.001)。该研究表明补体系统经典途径活化,并且支持慢性局部免疫过程参与该病发病机制的可能性(Mattila,J.等,Eur.Urol.9:350-352,1983)。而且,在自身抗体与膀胱粘膜中的抗原结合之后,补体活化可能参与组织损伤的产生,并参与该病典型的慢性自身延续性炎症(self-perpetuating inflammation)(Helin,H.等,Clin.Immunol.Immunopathol.43:88-96,1987)。
除了补体在泌尿生殖器炎性疾病中的作用之外,生殖功能也可能受到补体途径局部调节的影响。天然存在的补体抑制剂已经发展到了为宿主细胞提供所需的保护以控制体内的补体系统。为了揭示胚胎发育中的补体调控,对Crry进行了研究,Crry是天然存在的啮齿动物补体抑制剂,在结构上类似于人补体抑制剂MCP和DAF。有趣的是,产生Crry-/-小鼠的努力没有成功。相反,发现纯合的Crry-/-小鼠死于子宫内。Crry-/-胚胎在交配后存活到大约10天,存活随着由发育停滞所导致的死亡而迅速下降。炎性细胞也明显侵入Crry-/-胚胎的胎盘组织中。相比之下,Crry+/+胚胎似乎具有沉积在胎盘上的C3。这表明在胎盘水平上发生了补体活化,在缺少补体调节时,胚胎死亡。验证性研究在C3缺陷型背景下进行了引入Crry突变的试验。该补救性策略是成功的。总的说来,这些数据表示必须调节胎儿母体的补体界面。胎盘中补体调节的细微变化可能是胎盘功能障碍和流产的原因(Xu,C.等,Science 287:498-507,2000)。
先兆子痫是妊娠诱发的高血压症,其中涉及补体系统活化,不过仍然存在着争议(Haege,M.,Int.J.Gynecol.Obstet.43:113-127,1993)。体循环中的补体活化与先兆子痫中已确立的疾病密切相关,但是在临床症状出现之前没有发现补体活化增加,因此不能将补体成分用作先兆子痫的预测因子(Haeger等,Obstet.Gynecol.78:46,1991)。然而,在胎盘床局部环境中增加的补体活化可能胜于局部控制机制,导致过敏毒素和C5b-9水平升高(Haeger等,Obstet.Gynecol.73:551,1989)。
与抗精子抗体(ASA)相关的不育的一种假定机制是通过生殖道中补体活化的作用。C3b和iC3b调理素可通过其补体受体以及精子表面末端C5b-9复合物的形成来增强吞噬细胞与精子的结合,进而降低精子的活动性,故C3b和iC3b调理素的产生是与生殖力降低有关的可能原因。在不孕妇女的卵巢卵泡液中也证实了C5b-9水平升高(D'Cruz,O.J.等,J.Immunol.144:3841-3848,1990)。其它研究表明精子游动减缓,精子/卵子相互作用减弱,这些都可能与补体有关(D'Cruz,O.J.等,J.Immunol.146:611-620,1991;Alexander,N.J.,Fertil.Steril.41:433-439,1984)。最后,用sCR1进行的研究证实了针对ASA介导和补体介导的人精子损伤的保护作用(D'Cruz,O.J.等,Biol.Reprod.54:1217-1228,1996)。这些数据为补体抑制剂用于治疗泌尿生殖系统疾病和障碍提供了若干证据。
因此,本发明一方面提供用于抑制患有泌尿生殖系统疾病的患者的MASP-2依赖性补体活化的方法,该方法通过将包含治疗有效量的MASP-2抑制剂在药物载体中的组合物给予患有泌尿生殖系统疾病的受试者实施。我们认为用本发明方法和组合物进行治疗性治疗的泌尿生殖系统疾病以非限制性实例方式包括:疼痛性膀胱病、感觉性膀胱病、慢性非细菌性膀胱炎和间质性膀胱炎、男女不育、胎盘功能障碍以及流产和先兆子痫。可以全身性给予受试者MASP-2抑制剂,例如通过动脉内、静脉内、肌内、吸入、皮下或其它胃肠外给药,或者对于非肽能抑制剂可口服给予。或者,MASP-2抑制剂组合物可局部递送到泌尿生殖道,例如通过用液体溶液或凝胶组合物进行膀胱内冲洗或灌输。可按照医师规定重复给药以控制或消除疾病。
糖尿病和糖尿病性疾病
糖尿病性视网膜微血管病的特征是通透性增加、白细胞淤滞、微血栓形成和毛细血管细胞凋亡,所有这些都可能是由补体活化所引起或促进的。糖尿病患者的肾小球结构和神经内微血管都表现出补体活化的迹象。高含量葡萄糖体外选择性地降低CD55和CD59在内皮细胞表面上表达和CD59进行阻碍其补体抑制功能的非酶糖化作用的发现表明,糖尿病中补体抑制剂的利用度或有效性降低,CD55和CD59是糖基磷脂酰肌醇(GPI)锚定膜蛋白的两种抑制剂。
Zhang等人的研究(Diabetes51:3499–3504,2002)探索了作为人非增殖性糖尿病性视网膜病特征的补体活化及其与抑制分子变化的关联性。发现补体活化末端产物C5b-9的沉积发生在2型糖尿病病人眼供体的视网膜血管壁上,但在年龄匹配的非糖尿病供体血管则没有发生。在糖尿病性视网膜中未检测到经典途径特有的补体成分C1q和C4,这表明C5b-9是经替代途径产生的。糖尿病供体显示,由GPI锚连接到质膜上的两种补体抑制剂CD55和CD59的视网膜水平显著降低。在用10周链脲佐菌素诱发的糖尿病大鼠中观察到视网膜血管中类似的补体活化及视网膜CD55和CD59水平选择性降低。因此,糖尿病似乎引起补体抑制剂和补体活化的缺损性调节,其在糖尿病性视网膜微血管病大多数其它表现之前。
Gerl等人(Investigative Ophthalmology and Visual Science 43:1104-08,2000)测定了受累于糖尿病性视网膜病眼内的活化补体成分的存在。免疫组织化学研究表明,在所有50个糖尿病性视网膜病样本中,在紧邻玻璃膜(Bruch membrane)之下以及紧包住毛细血管的脉络膜毛细血管层中都检测到了补体C5b-9复合物大量沉积。C3d染色与C5b-9染色正相关,表示补体活化在原位发生的事实。此外,发现了玻连蛋白的阳性染色,它与胞外C5b-9形成了稳定的复合物。相反,没有C反应蛋白(CRP)、甘露聚糖结合凝集素(MBL)、C1q或C4的阳性染色,表明补体活化不是通过C4依赖性途径发生的。因此,C3d、C5b-9和玻连蛋白的存在表明,补体活化可能是通过糖尿病性视网膜病受累眼中脉络膜毛细血管层中的替代途径而完成的。补体活化可能是可造成眼组织疾病和视觉缺陷的病理性后遗症的病因。因此,使用补体抑制剂可能是减少或阻断糖尿病中发生的微血管损伤的有效治疗方法。
胰岛素依赖型糖尿病(IDDM,亦称I型糖尿病)是与不同类型自身抗体的存在有关的自身免疫性疾病(Nicoloff等,Clin.Dev.Immunol.11:61-66,2004)。这些抗体和相应抗原在循环中的存在导致形成循环免疫复合物(CIC),已知CIC长期保留在血液中。CIC在小血管中的沉积有可能导致伴有衰弱性临床结果的微血管病。糖尿病儿童中,CIC与微血管并发症的发生之间存在关联性。这些发现表明CIC IgG的水平升高与早期糖尿病性视网膜病的发生有关,且补体途径抑制剂对于阻断糖尿病性视网膜病可能是有效的(Kotnik等,Croat.Med.J.44:707-11,2003)。此外,下游补体蛋白的形成和替代途径的参与可能是IDDM中胰岛细胞总体功能的影响因素,期望使用补体抑制剂来减少可能的损伤或限制细胞死亡(Caraher等,J.Endocrinol.162:143-53,1999)。
与健康对照相比,循环MBL浓度在1型糖尿病患者中显著升高,并且这些MBL浓度与尿白蛋白排泄正相关(Hansen等,J.Clin.Endocrinol.Metab.88:4857-61,2003)。最近的临床研究发现,MBL基因型高表达和低表达的频率在1型糖尿病患者和健康对照之间是相似的(Hansen等,Diabetes53:1570-76,2004)。然而,如果糖尿病患者的MBL基因型高,则他们患肾病的风险显著提高。这就表明MBL高水平和凝集素途径补体活化可能是引起糖尿病性肾病发生的原因。该结论得到最新前瞻性研究的支持,在该研究中,对MBL水平与新近诊断为 1型糖尿病患者组群中蛋白尿发生之间的关联性进行了分析(Hovind等,Diabetes54:1523-21,2005)。他们发现在1型糖尿病病程早期,高水平的MBL与稍后持续性蛋白尿的发生显著相关。这些结果表明,MBL和凝集素途径可能参与糖尿病血管并发症的特定发病机制,而不仅仅是引起现有病变的速度加快。在最近的临床研究(Hansen等,Arch.Intern.Med.166:2007-13,2006)中,在接受随访调查15年以上,已进行基线充分鉴定的一组2型糖尿病患者中,对MBL水平进行了测定。他们发现即使在对已知的混淆变量进行调整后,MBL血浆水平高(>1000μg/L)的患者的死亡风险比低水平MBL(<1000μg/L)患者明显较高。
本发明另一方面,提供抑制患有非肥胖性糖尿病(IDDM)或IDDM的血管病、神经病或视网膜病并发症或者成人发病的(2型)糖尿病的受试者的MASP-2依赖性补体活化的方法,该方法通过将包含治疗有效量的MASP-2抑制剂在药物载体中的组合物给予受试者实施。可以全身性给予受试者MASP-2抑制剂,例如通过动脉内、静脉内、肌内、皮下或其它胃肠外给药,或者对于非肽能抑制剂可口服给予。或者,可通过局部送递到血管病、神经病或视网膜病症状的部位来给药。可在一段长的时间内定期给予MASP-2抑制剂以治疗或控制慢性病,或者通过单次或连续给药来治疗急性病。
围化疗期的给药和恶性肿瘤的治疗
补体系统的活化还可能参与恶性肿瘤的发病机制。最近,使用多克隆抗体或单克隆抗体以及链霉抗生物素-生物素-过氧化物酶技术,对17个乳腺癌样品和6个良性乳腺肿瘤样品中的C5b-9补体复合物新抗原、IgG、C3、C4、S蛋白/玻连蛋白、纤连蛋白和巨噬细胞进行了定位。所有的癌组织样品在每个TNM阶段,都有C5b-9沉积在肿瘤细胞膜上,有细颗粒沉积在细胞残留物上,并且坏死区域中出现弥散性沉积物(Niculescu,F.等,Am.J.Pathol.140:1039-1043,1992)。
此外,补体活化可能是化学疗法或放射治疗的结果,因此抑制补体活化可能用作恶性肿瘤治疗的辅助治疗以减少医源性炎症。手术之前进行化学治疗和放射治疗时,C5b-9的沉积更多更久。在所有良性病变样品中都不存在C5b-9沉积物。S蛋白/玻连蛋白呈原纤维状沉积物存在于结缔组织基质中,呈弥散性沉积物存在于肿瘤细胞周围,不如纤连蛋白沉积得多而持久。IgG、C3和C4沉积物只存在于癌样品中。乳腺癌中C5b-9沉积物的存在表示补体活化及其随后的致病效应(Niculescu,F.等,Am.J.Pathol.140:1039-1043,1992)。
脉冲式可调谐染料激光(577nm)(PTDL)治疗诱导了血红蛋白凝固和组织坏死,这主要限于血管中。在经PTDL照射的正常皮肤的研究中,主要发现如下:1)C3片段、C8、C9和MAC沉积在血管壁上;2)这些沉积物不是由于蛋白变性,因为它们仅仅在照射后7分钟就很明显了,与运铁蛋白立即沉积在红细胞凝固部位上不同;3)显示C3沉积物通过替代途径放大了补体活化,这是特异性的反应,因为组织坏死本身并不导致这种放大作用;和4)这些反应是在多形核白细胞局部累积之前发生的。在血管瘤中组织坏死更明显。坏死中心的较大血管瘤血管并不显著地固定补体。相反,位于外周血管中的补体沉积类似于正常皮肤中所观察到的沉积,除了一个例外:在激光治疗后立即在一些血管中检测到C8、C9和MAC,这个发现与没有C5转化酶形成而直接发生MAC装配相一致。这些结果表明,补体在PTDL诱导的血管坏死中被激活,并可能是随后的炎症反应的原因。
肿瘤的光动力学疗法(PDT)引起了强烈的宿主免疫应答,其表现之一是显著的嗜中性粒细胞增多。除了由于PDT诱导的补体活化而释放的补体片段(直接介质)以外,还有至少12种由于补体活性而引起的间接介质。后者包括细胞因子IL-1β、TNF-α、IL-6、L-10、G-CSF和KC、血栓烷、前列腺素、白三烯、组胺和凝血因子(Cecic,I.等,Cancer Lett.183:43-51,2002)。
最后,可以展望MASP-2依赖性补体活化的抑制剂与标准治疗方案联用以治疗癌症。例如,用利妥昔单抗(rituximab),即一种嵌合抗CD20单克隆抗体进行治疗可能与中度至严重的首剂不利作用相关,特别是在具有大量循环肿瘤细胞的患者中。在最近的首次输注利妥昔单抗的研究中,在5位复发的轻度非霍奇金淋巴瘤(NHL)患者中,测得补体活化产物(C3b/c和C4b/c)和细胞因子(肿瘤坏死因子α(TNF-α)、白介素6(IL-6)和IL-8)。输注利妥昔单抗诱导快速的补体活化,其先于TNF-α、IL-6和IL-8的释放。尽管研究组小,但是补体活化水平似乎是与输注前的循环B细胞数量有关(r=0.85;P=0.07),并与副作用的严重性有关。这些结果表明,补体在利妥昔单抗治疗副作用的发生机制中起着关键作用。因为不能用皮质类固醇防止补体活化,所以研究补体抑制剂在利妥昔单抗首次用药期间的可能作用中具有重大意义(van der Kolk,L.E.等,Br.J.Haematol.115:807-811,2001)。
在本发明另一个方面,提供用于抑制待接受化学疗法和/或放射疗法治疗的受试者的MASP-2依赖性补体活化的方法,所述疗法包括但不限于癌症状况治疗。该方法包括将包含治疗有效量的MASP-2抑制剂在药物载体中的组合物在围化疗期,即在实施化学疗法和/或放射疗法之前和/或期间和/或之后给予患者。例如,可以在实施化学疗法或放射疗法之前或者实施化学疗法或放射疗法同时给予本发明的MASP-2抑制剂组合物,并在整个治疗过程中持续,以减轻化学疗法和/或放射疗法在非目标健康组织中的有害影响。此外,可以在化学疗法和/或放射疗法之后给予MASP-2抑制剂组合物。要理解的是,化学疗法和放射疗法方案经常需要重复治疗,因此MASP-2抑制剂组合物的给药也可重复,并与化学疗法和放射疗法相对一致。还认为MASP-2抑制剂可用作化疗药物,单用或与其它化疗药物和/或放射疗法联用,以治疗患有恶性肿瘤的患者。可以适当通过口服(用于非肽能药物)、静脉内、肌内或其它非肠道途径给药。
内分泌疾病
最近还发现与少数几种内分泌疾病或失调有关的补体系统,包括桥本甲状腺炎(Hashimoto's thyroiditis)(Blanchin,S.等,Exp.Eye Res.73(6):887-96,2001)、应激、焦虑症以及其它涉及自垂体调节性释放的催乳素、生长因子或胰岛素样生长因子和促肾上腺皮质激素的潜在的激素失调(Francis,K.等,FASEB J.17:2266-2268,2003;Hansen,T.K.,Endocrinology144(12):5422-9,2003)。
使用例如激素和细胞因子这类分子,在内分泌系统和免疫系统之间形成了双向沟通。最近,阐明了一条新途径,通过该途径,补体衍生的细胞因子C3a刺激垂体前叶激素释放并激活对应激反应和炎症控制的反射中枢下丘脑-垂体-肾上腺轴。C3a受体在分泌垂体激素的细胞和不分泌垂体激素的(滤泡星形(folliculostellate))细胞中表达。C3a和脱Arg的C3a(非炎性代谢物)刺激垂体细胞培养物释放催乳素、生长激素和促肾上腺皮质激素。体内给予重组C3a和脱Arg的C3a时,这些激素以及肾上腺皮质酮的血清水平以剂量依赖性方式增加。这就表明补体途径通过与内分泌垂体腺的沟通来调节组织特异性和全身性炎症反应(Francis,K.等,FASEB J.17:2266-2268,2003)。
越来越多的动物和人体研究表明,生长激素(GH)和胰岛素样生长因子I(IGF-I)调节免疫功能。GH疗法增加垂危患者的死亡率。过量的死亡几乎完全是由败血症性休克或多器官衰竭所致,这表明可能涉及GH诱导的免疫和补体功能的调节。甘露聚糖结合凝集素(MBL)是在先天免疫中通过结合糖结构之后激活补体级联和炎症而起重要作用的血浆蛋白。有证据支持,生长激素对MBL水平有重要影响,因此可能对凝集素依赖性补体活化有重要影响(Hansen,T.K.,Endocrinology 144(12):5422-9,2003)。
甲状腺过氧化物酶(TPO)是参与自身免疫性甲状腺疾病的一种主要的自身抗原。TPO由大的N末端髓过氧化物酶样模块(module)、其后是补体调控蛋白(CCP)样模块和表皮生长因子样模块组成。CCP模块是参与C4补体成分活化的分子组分,在自身免疫性疾病中研究C4能否结合TPO并激活补体途径。TPO通过其CCP模块直接激活补体,而无需Ig的任何介导。而且,在桥本甲状腺炎患者中,甲状腺细胞过量表达C4和该补体途径的所有下游成分。这些结果表明,TPO连同与补体途径活化有关的其它机制一起,可能是桥本甲状腺炎中观察到的大量细胞遭破坏的原因(Blanchin,S.等,2001)。
因此,本发明一方面提供用于抑制MASP-2依赖性补体活化以治疗内分泌疾病的方法,该方法通过将包含治疗有效量的MASP-2抑制剂在药物载体中的组合物给予患有内分泌疾病的受试者实施。根据本发明进行治疗的疾病以非限制性实例方式包括:桥本甲状腺炎、应激、焦虑症以及其它涉及自垂体调节性释放的催乳素、生长因子或胰岛素样生长因子和促肾上腺皮质激素的潜在的激素失调。可全身性给予受试者MASP-2抑制剂,例如通过动脉内、静脉内、肌内、吸入、经鼻、皮下或其它胃肠外给药,或者对于非肽能抑制剂可口服给予。MASP-2抑制剂组合物可以与一种或多种另外的治疗药物联用。可按照医师规定重复给药直到疾病消退。
眼科疾病
年龄相关性黄斑变性(AMD)是累及数百万成人的致盲疾病,然而对导致AMD发生的生物化学、细胞和/或分子事件的后遗症了解甚少。AMD导致黄斑的渐进性破坏,这与位于黄斑内及其周围、视网膜后以及视网膜色素上皮(RPE)与脉络膜之间的称为脉络膜小疣(drusen)的胞外沉积物的形成有关。最近的研究揭示,与炎症和免疫介导过程相关的蛋白质在脉络膜小疣相关成分中是普遍的。已经在视网膜、RPE和脉络膜细胞检测到编码许多这类分子的转录物。这些数据还证实,作为有效抗原呈递细胞的树突细胞与脉络膜小疣的发生密切相关,补体活化是脉络膜小疣内部以及沿着RPE–脉络膜界面的关键途径(Hageman,G.S.等,Prog.Retin.Eye Res.,20:705-732,2001)。
若干项独立研究表明,AMD与补体H因子(CFH)基因的遗传多态性之间有强关联性,其中在危险性等位基因纯合的个体中AMD的可能性增加了7.4倍(Klein,R.J.等,Science,308:362-364,2005;Haines等,Science 308:362-364,2005;Edwards等,Science308:263-264,2005)。CFH基因已经被定位在染色体1q31,这是一个在AMD中涉及6个独立连锁扫描(linkage scan)的区域(参见例如Schultz,D.W.等,Hum.Mol.Genet.12:3315,2003)。已知CFH是补体系统的关键调节因子。研究显示,细胞上和循环中的CFH通过抑制将C3激活成为C3a和C3b以及通过钝化现有的C3b来调节补体活性。已经在AMD患者的玻璃膜(Brusch's membrane)、毛细血管间支柱(intercapillary pillar)中以及脉络膜小疣内部观察到C5b-9的沉积(Klein等)。免疫荧光实验表明,在AMD中CFH的多态性可能引起补体沉积在脉络膜毛细血管和脉络膜血管中(Klein等)。
膜结合补体抑制剂补体受体1也定位在脉络膜小疣中,但是在RPE细胞中用免疫组织化学法无法检测出。相比之下,第二膜结合补体抑制剂膜膜辅因子蛋白存在于脉络膜小疣结合的RPE细胞中以及脉络膜小疣内部的小的球形亚结构组分中。这些以前未鉴定出的组分对特征性沉积在补体活化部位的补体成分C3的蛋白水解片段也具有强的免疫反应性。有研究认为这些结构代表来自作为补体攻击目标的退行性RPE细胞的残余碎片(Johnson,L.V.等,Exp.Eye Res.73:887-896,2001)。
这些多补体调节剂以及补体活化产物(C3a、C5a、C3b、C5b-9)的鉴定和定位使研究者断定,慢性补体活化在脉络膜小疣生物发生过程和AMD病因中起着重要作用(Hageman等,Progress Retinal Eye Res.20:705-32,2001)。在脉络膜小疣中鉴定出C3和C5活化产物,并不会让人认了解补体是通过经典途径还是通过凝集素途径或替代放大环路活化,正如本发明所了解的一样,因为C3和C5两者是所有三条途径共有的。然而,两项研究使用对经典途径活化必不可少的识别成分C1q特异性的抗体,对脉络膜小疣免疫标记进行了探索(Mullins等,FASEB J.14:835-846,2000;Johnson等,Exp.Eye Res.70:441-449,2000)。两项研究都得出结论,认为在脉络膜小疣中一般观察不到C1q免疫标记。这些有关C1q的否定结果表明,脉络膜小疣中的补体活化不是通过经典途径产生的。另外,Mullins等人的研究(2000)报道了脉络膜小疣的免疫复合物组分(IgG轻链、IgM)的免疫标记是微弱可变的,这进一步表明经典途径在该疾病过程期间发生的补体活化中起着次要作用。
最近,两篇已发表的研究报告对小鼠(一种人CNV模型)中补体在激光诱导的脉络膜新血管形成(CNV)的发展中的作用进行了评价。Bora及其同事(2005)采用免疫组织学方法发现,在激光处理后,补体活化产物C3b和C5b-9(MAC)在新血管复合体上的显著沉积(Bora等,J.Immunol.174:491-7,2005)。重要的是,在C3遗传缺陷型小鼠(C3-/-小鼠)中不发生CNV,C3是所有补体活化途径所需的必不可少的成分。在激光诱导的CNV后,对小鼠眼组织中的3种涉及CNV的血管生成因子VEGF、TGF-β2和β-FGF的RNA信息水平进行了评价。值得注意的是,补体缺失导致这些血管生成因子的RNA水平明显降低。
Nozaki及其同事采用ELISA方法,证实了在激光诱导的CNV进程的早期产生了有效的过敏毒素C3a和C5a(Nozaki等,Proc.Natl.Acad.Sci.U.S.A.103:2328-33,2006)。此外,在野生型小鼠中,将C3和C5的这两种生物活性片段注射到玻璃体腔之后诱导VEGF表达。与这些结果一致,Nozaki及其同事还表明遗传上去除C3a和C5a的受体减少激光损伤后的VEGF表达和CNV形成,抗体介导的C3a或C5a中和或其受体的药理阻断也减少CNV。之前的研究证实,白细胞募集,特别是巨噬细胞募集在激光诱导的CNV中起着关键作用(Sakurai等,Invest.Opthomol.Vis.Sci.44:3578-85,2003;Espinosa-Heidmann等,Invest.Opthomol.Vis.Sci.44:3586-92,2003)。在Nozaki及其同事的论文(2006)中报道了在激光损伤后,C3aR(-/-)和C5aR(-/-)小鼠的白细胞募集显著减少。
因此,本发明的一方面提供用于抑制MASP-2依赖性补体活化以治疗年龄相关性黄斑变性或其它补体介导的眼科疾病的方法,该方法通过将包含治疗有效量的MASP-2抑制剂在药物载体中的组合物给予患有这些疾病或其它补体介导的眼科疾病的受试者实施。可通过例如凝胶剂、软膏剂或滴剂形式的组合物经冲洗或敷用而局部给予眼部MASP-2抑制剂组合物。或者,可全身性给予受试者MASP-2抑制剂,例如通过动脉内、静脉内、肌内、吸入、经鼻、皮下或其它胃肠外给药,或者对于非肽能抑制剂可口服给予。MASP-2抑制剂组合物可与一种或多种另外治疗药物联用,另外的治疗药物公开于例如美国专利申请公开号2004-0072809-A1。可按照医师规定重复给药直到疾病消退或得到控制。
凝血病
已形成补体系统在弥散性血管内凝血("DIC")如显著身体外伤继发的DIC中的作用的证据。
前述研究已显示,C4-/-小鼠不受免于肾再灌注损伤的保护(Zhou,W.等人,"Predominant role for C5b-9in renal ischemia/reperfusion injury,"J Clin Invest 105:1363-1371(2000))。为了研究C4-/-小鼠是否仍然能够经由经典或凝集素途径激活补体,在特异性针对经典或凝集素途径激活路线的试验中测定C4-/-血浆中的C3周转。尽管在经由经典途径触发激活时未能观察到C3裂解,但观察到C4缺陷血清中的C3的高效凝集素途径依赖性激活(图30)。能够看出,在MASP-2-/-小鼠体内,甘露聚糖和酵母聚糖上的C3b沉积严重受损,即使在根据许多先前发表的关于替代途径激活的论文,应该对于所有三条途径而言是允许的实验条件下。当使用由免疫球蛋白复合体包被而不是甘露聚糖或酵母聚糖包被的孔中的相同血清时,在MASP-2+/+小鼠血清和MASP-2-/-血清中看到C3b沉积和因子B裂解,而在C1q耗尽的血清中看不到。这表明,当经由经典活性提供初始的C3b时,在MASP-2-/-血清中促进了替代途径激活。图30C图示以下令人惊讶的发现:C3能够在C4缺陷血浆中以凝集素途径依赖的形式被有效激活。
这个“C4旁路”通过经由用可溶性甘露聚糖或甘露糖预温育血浆的凝集素途径激活的抑制而废止。
异常、非免疫性的补体系统的激活对人是具有潜在危险性的,并且可能还在血液途径激活中起重要作用,尤其是在其中炎性途径和血液途径均被激活的严重外伤的情况下。在正常的健康者体内,C3转化率是总血浆C3蛋白的<5%。在猛烈的感染(包括败血症和免疫复合物病)中,C3转化率在补体水平常常低于正常水平的情况下以约30%重建自身,这归因于增加的利用和池(pool)分布的改变。大于30%的即时C3途径激活通常产生血管舒张和组织体液丧失的明显的临床证据。在30%C3转化率以上,引发机制主要是非免疫性的,并且导致的临床表现对患者有害。健康者体内和对照患病者体内的补体C5水平似乎远比C3更稳定。C5水平的显著降低和/或转化与患者对非正常多发伤(例如,道路交通事故)和可能形成休克肺综合症的反应相关联。因此,超过30%的血管池的补体C3激活的任何证据、或任何C5介入的任何证据、或两者可被认为可能是患者中有害病理变化的前兆。
C3和C5两者均放出作用于释放血管舒张化合物的肥大细胞和嗜碱细胞的过敏毒素(C3a和C5a)。它们建立趋化性梯度以将多形核细胞(PMN)导向免疫干扰(一种有益的应答)的中心,但在这里它们不一样,因为C5a对这些吞噬细胞具有特异性凝块(凝集)作用,防止它们远离反应位点的随机移动。在感染的正常对照中,C3激活C5。然而,在多发伤中,C5似乎被广泛激活,系统地生成C5a过敏毒素。此不受控制的活性引起多形白细胞在血管系统内部凝块,并且然后这些凝块被扫入肺的毛细血管,它们由于过氧化物的放出而在其中滞留并且产生局部损伤作用。尽管不愿被理论限制,但该机制在急性呼吸窘迫综合征(ARDS)的发病机理中或许是重要的,不过此见解最近受到了质疑。在体外,C3a过敏毒素能够显示为强有力的血小板凝聚体,但它们的体内介入较少被定义,并且伤口修复中的血小板物质和纤溶酶的释放可仅次要地包含补体C3。可能的是,C3激活的延时上升为生成DIC所必需。
除以上概述的能够解释外伤与DIC之间的联系的被激活补体成分的细胞和血管效应之外,出现的科学发现识别了补体与凝血系统之间的直接分子连接和功能性交互。支持数据已从对C3缺陷小鼠的研究中获得。由于C3是各补体途径的共享成分,所以预测C3缺陷小鼠缺乏全部补体功能。然而,意外地,C3缺陷小鼠能够完美激活终端补体成分(Huber-Lang,M.等人,"Generation of C5a in the absence of C3:a new complement activation pathway,"Nat.Med 12:682-687(2006))。研究深入揭示,终端补体成分的C3依赖性激活是通过凝血酶(凝血级联的限速酶)介导的(Huber等人,2006)。初始补体活化之后介导凝血酶激活的分子成分仍然是难以捉摸的。
本发明人阐明了什么被认为是补体与凝血级联之间的交互的分子基础,并且将MASP-2识别为连接该两个系统的中心控制点。除众所周知的C2和C4补体蛋白之外,对MASP-2的底物特异性的生化研究已将凝血酶原鉴定为可能的底物。MASP-2在功能相关的位点特异性裂解凝血酶原,生成凝血酶,即凝血级联的限速酶(Krarup,A.等人,"Simultaneous Activation of Complement and Coagulation by MBL-Associated Serine Protease 2,"PLoS.ONE.2:e623(2007))。由MASP-2生成的凝血酶能够促进纤维蛋白在限定的重建的体外系统中的沉积,证明MASP-2裂解的功能相关性(Krarup等人,2007)。如以下本文实例中所论述,发明人通过证明凝集素途径激活之后正常啮齿动物血清内的凝血酶激活,进一步印证了这个发现的生理学意义,并且证明了这个过程通过中和MASP-2单克隆抗体而被阻断。
MASP-2可在凝集素途径中代表中心分支点,能够促进补体系统和凝血系统两者的激活。由于凝集素途径激活是对许多类型的外伤性损伤的生理应答,所以本发明人认为,并发的(由补体成分介导的)系统性炎症和(经由凝血途径介导的)弥散性凝血能够通过MASP-2激活两个途径的能力来解释。这些发现清楚地表明MASP-2在DIC产生中的作用和MASP-2抑制在治疗或预防DIC中的治疗学益处。MASP-2可提供补体系统与凝血系统之间的分子连接,并且凝集素途径的激活在其发生在外伤的背景下时能够经由MASP-2凝血酶轴直接引发凝血系统的激活,提供外伤和DIC之间的机制联系。根据本发明的一个方面,MASP-2的抑制会抑制凝集素途径激活并且减少过敏毒素C3a和C5a两者的生成。据信,C3激活的延时上升为生成DIC所必需。
所以,本发明的一个方面因而提供用于抑制MASP-2依赖性补体活化以治疗弥散性血管内凝血或其他补体介导的凝血障碍的方法,该方法通过向患有此类病症或处于发生此类病症风险中的受试者给予组合物来进行,该组合物包含药物载体中的治疗有效量的MASP-2抑制剂(例如,抗MASP-2抗体或其片段、肽抑制剂或小分子抑制剂)。在一些实施方案中,MASP-2抑制剂能够阻断已被激活的MASP-2。将MASP-2抑制组合物适当给予受试者全身,诸如通过动脉内、静脉内、肌内、吸入、鼻、皮下或其他肠胃外给药,或对于非肽能剂而言可能通过口服给药。给药可按照医师决定而重复,直到已消除或控制病症为止。本发明的这个方面的方法可用于治疗DIC,其继发于败血症、包括神经性创伤(例如,急性颅脑损伤,见Kumura,E.等人,Acta Neurochirurgica85:23-28(1987))的严重外伤、感染(细菌、病毒、真菌、寄生虫)、癌症、产科并发症、肝病、严重中毒反应(例如,蛇咬伤、昆虫咬伤、输血反应)、休克、中暑、移植排异反应、血管动脉瘤、肝功能衰竭、通过化学治疗或放射治疗的癌症疗法、烧伤、意外辐射暴露及其他病因。见(例如)Becker J.U.和Wira C.R."Disseminated Intravascular Coagulation"emedicine.medscape.com/9/10/2009。对于外伤或其他急性事件继发的DIC而言,MASP-2抑制组合物可在外伤性损伤之后立即给药,或在外伤诱发损伤或状况(诸如对被认为处于DIC风险中的患者的手术)之前、期间、紧接着或之后一至七天或更长时间内(诸如在24小时至72小时内)给药。在一些实施方案中,MASP-2抑制组合物可以速效剂型适当给药,诸如通过包含MASP-2抑制剂组合物的溶液滴注剂的静脉内或动脉内输送。
IV.MASP-2抑制剂
一方面,本发明提供抑制MASP-2依赖性补体活化的不利作用的方法。MASP-2抑制剂是以有效抑制有生命的受试者的MASP-2依赖性补体活化的量给予的。在本发明这个方面的实施中,代表性的MASP-2抑制剂包括:抑制MASP-2生物活性的分子(例如小分子抑制剂、抗MASP-2抗体或者与MASP-2相互作用或妨碍蛋白间相互作用的封闭性肽),以及减少MASP-2表达的分子(例如MASP-2反义核酸分子、MASP-2特异性RNAi分子和MASP-2核酶),从而阻止MASP-2激活替代补体途径。MASP-2抑制剂可单独使用作为主要疗法,或者与其它治疗药物联用作为辅助疗法以增强其它药物治疗的治疗益处。
MASP-2依赖性补体活化抑制以补体系统成分的至少一种以下的变化为特征,所述变化是由于根据本发明的方法给予MASP-2抑制剂所致:MASP-2依赖性补体活化系统产物C4b、C3a、C5a和/或C5b-9(MAC)形成或产生的抑制(测定法参见例如实施例2)、使用未致敏兔或豚鼠红细胞在溶血分析中评价的替代补体活化的减少、C4裂解和C4b沉积的减少(测定法参见例如实施例2),或者C3裂解和C3b沉积的减少(测定法参见例如实施例2)。
根据本发明,使用有效抑制MASP-2依赖性补体活化系统的MASP-2抑制剂。用于实施本发明这个方面的MASP-2抑制剂包括例如抗MASP-2抗体及其片段、MASP-2抑制肽、小分子、MASP-2可溶性受体和表达抑制剂。MASP-2抑制剂可通过阻断MASP-2的生物学功能而抑制MASP-2依赖性补体活化系统。例如,抑制剂可有效阻断MASP-2蛋白间的相互作用,阻碍MASP-2二聚化或装配,阻断Ca2+结合,阻碍MASP-2丝氨酸蛋白酶活性位点,或者可减少MASP-2蛋白表达。
在一些实施方案中,MASP-2抑制剂选择性抑制MASP-2补体活化,保持了C1q依赖性补体活化系统功能上的完整性。
在一个实施方案中,用于本发明方法的MASP-2抑制剂是特异性的MASP-2抑制剂,其与包含SEQ ID NO:6的多肽特异性结合的亲和力是与补体系统中的其它抗原结合的亲和力的至少10倍高。在另一个实施方案中,MASP-2抑制剂与包含SEQ ID NO:6的多肽特异性结合的结合亲和力是与补体系统中的其它抗原结合的结合亲和力的至少100倍高。MASP-2抑制剂的结合亲和力可使用合适的结合测定法进行测定。
MASP-2多肽具有类似于C1补体系统的蛋白酶MASP-1、MASP-3以及C1r和C1s的分子结构。SEQ ID NO:4所示的cDNA分子编码MASP-2的代表性实例(由SEQ ID NO:5所示的氨基酸序列组成),提供带有前导序列(氨基酸1-15)的人MASP-2多肽,分泌后被切割,产生成熟形式的人MASP-2(SEQ ID NO:6)。如图2中所示,人MASP 2基因包括十二个外显子。人MASP-2cDNA由外显子B、C、D、F、G、H、I、J、K和L编码。可变剪接产生了20kDa蛋白,称为MBL相关的蛋白19(“MAp19”,亦称“sMAP”)(SEQ ID NO:2),由图2所示外显子B、C、D和E产生的(SEQ ID NO:1)编码。SEQ ID NO:50所示的cDNA分子编码鼠MASP-2(由SEQ ID NO:51所示的氨基酸序列组成),提供带有前导序列的鼠MASP-2多肽,分泌后被切割,产生成熟形式的鼠MASP-2(SEQ ID NO:52)。SEQ ID NO:53所示的cDNA分子编码大鼠MASP-2(由SEQ ID NO:54所示的氨基酸序列组成),提供带有前导序列的大鼠MASP-2多肽,分泌后被切割,产生成熟形式的大鼠MASP-2(SEQ ID NO:55)。
本领域技术人员应理解的是,SEQ ID NO:4、SEQ ID NO:50和SEQ ID NO:53中公开的序列分别代表人、小鼠和大鼠MASP-2的单个等位基因,发生等位基因变异和可变剪接是预料之中的。SEQ ID NO:4、SEQ ID NO:50和SEQ ID NO:53所示核苷酸序列的等位基因变体,包括含有沉默突变的变体以及其中导致氨基酸序列改变的突变的变体,都属本发明的范围。可根据标准方法,通过探测cDNA或基因组文库而从不同个体中克隆MASP-2序列的等位基因变体。
人MASP-2蛋白(SEQ ID NO:6)的结构域见图3A,包括N端C1r/C1s/海胆Vegf/骨形成蛋白(CUBI)结构域(SEQ ID NO:6的氨基酸1-121)、表皮生长因子样结构域(氨基酸122-166)、第二CUBI结构域(氨基酸167-293)以及串联的补体调控蛋白结构域和丝氨酸蛋白酶结构域。MASP 2基因的可变剪接产生图3B中所示的MAp19。MAp19是含有MASP-2的N端CUB1-EGF区的非酶蛋白,具有得自图2所示外显子E的4个额外残基(EQSL)。
研究表明,一些蛋白质通过蛋白间的相互作用与MASP-2结合或者与MASP-2相互作用。例如,已知MASP-2结合凝集素蛋白MBL、H-纤维胶凝蛋白和L-纤维胶凝蛋白,并与之形成Ca2+依赖性复合物。研究表明,每种MASP-2/凝集素复合物通过蛋白质C4和C2的MASP-2依赖性裂解来激活补体(Ikeda,K.等,J.Biol.Chem.262:7451-7454,1987;Matsushita,M.等,J.Exp.Med.176:1497-2284,2000;Matsushita,M.等,J.Immunol.168:3502-3506,2002)。研究表明,MASP-2的CUB1-EGF结构域对于MASP-2与MBL的缔合是必不可少的(Thielens,N.M.等,J.Immunol.166:5068,2001)。研究还表明,CUB1EGFCUBII结构域介导MASP-2的二聚化,这是形成活性MBL复合物所需要的(Wallis,R.等,J.Biol.Chem.275:30962-30969,2000)。因此,可对结合已知对MASP-2依赖性补体活化重要的MASP-2靶区或者阻碍该区域的MASP-2抑制剂进行鉴定。
抗MASP-2抗体
在本发明这个方面的一些实施方案中,MASP-2抑制剂包括抑制MASP-2依赖性补体活化系统的抗MASP-2抗体。用于本发明这个方面的抗MASP-2抗体包括得自任何产生抗体的哺乳动物的多克隆抗体、单克隆抗体或重组抗体,并且可以是多特异性的、嵌合的、人源化的、抗独特型抗体以及抗体片段。抗体片段包括Fab、Fab'、F(ab)2、F(ab')2、Fv片段、scFv片段和单链抗体,见本文的进一步描述。
文献中记载了几种抗MASP-2抗体,其中的一些列于下表1。可使用本文所述测定法筛选前述抗MASP-2抗体抑制MASP-2依赖性补体活化系统的能力。例如,已经鉴定出阻断MASP-2依赖性补体活化的抗大鼠MASP-2Fab2抗体,详情见本文实施例24和25。一旦鉴定出发挥MASP-2抑制剂作用的抗MASP-2抗体,就可将它用于产生抗独特型抗体,并用于鉴定其它MASP-2结合分子,如见下文进一步描述的。
表1:来自文献的MASP-2特异性抗体
效应子功能降低的抗MASP-2抗体
在本发明这个方面的一些实施方案中,抗MASP-2抗体降低了效应子功能,来减轻可能由经典补体途径活化产生的炎症。研究表明IgG分子引发经典补体途径的能力存在于分子的Fc部分(Duncan,A.R.等,Nature 332:738-740 1988)。其中通过酶切割除去分子Fc部分的IgG分子缺少这种效应子功能(参见Harlow,Antibodies:A Laboratory Manual,Cold Spring Harbor Laboratory,New York,1988)。因此,可通过具有使效应子功能减到最低的基因工程Fc序列,或者成为人IgG2或IgG4同种型,由此去掉分子的Fc部分,从而产生效应子功能降低的抗体。
可对IgG重链的Fc部分进行标准分子生物学操作来产生效应子功能降低的抗体,如本文实施例9所述,也描述于Jolliffe等,Int’l Rev.Immunol.10:241-250,1993和Rodrigues等,J.Immunol.151:6954-6961,1998。效应子功能降低的抗体还包括激活补体和/或与Fc受体相互作用的能力降低的人IgG2和IgG4同种型(Ravetch,J.V.等,Annu.Rev.Immunol.9:457-492,1991;Isaacs,J.D.等,J.Immunol.148:3062-3071,1992;van de Winkel,J.G.等,Immunol.Today 14:215-221,1993)。可通过本领域普通技术人员已知的若干方法之一,来产生对包括IgG2或IgG4同种型在内的人MASP-2特异性的人源化抗体或完全人抗体,如Vaughan,T.J.等,Nature Biotechnical 16:535-539,1998所述。
抗MASP-2抗体的产生
可使用MASP-2多肽(例如全长MASP-2)或使用带有抗原性MASP-2表位的肽(如MASP-2多肽部分)来产生抗MASP-2抗体。免疫原性肽可以少至五个氨基酸残基。例如,可以使用包括SEQ ID NO:6全部氨基酸序列的MASP-2多肽来诱导用于本发明方法的抗MASP-2多肽。可以将已知参与蛋白间相互作用的特定MASP-2结构域(例如CUBI)和CUBIEGF结构域以及包括丝氨酸蛋白酶活性部位的区域表达为实施例5中所述的重组多肽并用作抗原。此外,包含MASP-2多肽(SEQ ID NO:6)至少6个氨基酸的部分的肽也可用来诱导MASP-2抗体。下表2中提供了用于诱导MASP-2抗体的MASP-2衍生抗原的其它实例。用于产生抗体的MASP-2肽和多肽可作为天然多肽、或者重组肽或合成肽以及无催化活性的重组多肽(例如MASP-2A)而被分离出来,如实施例5-7进一步描述的。在本发明这个方面的一些实施方案中,使用实施例8和实施例9中所述转基因小鼠品系获得抗MASP-2抗体,如下文进一步描述的。
用于产生抗MASP-2抗体的抗原还包括融合多肽,例如MASP-2或其部分与免疫球蛋白多肽或者与麦芽糖结合蛋白的融合物。多肽免疫原可以是全长分子或其部分。如果多肽部分是半抗原样的,则最好可将这部分结合或连接到大分子载体(例如匙孔血蓝蛋白(KLH)、牛血清白蛋白(BSA)或破伤风类毒素)上用于免疫。
表2:MASP-2衍生的抗原
多克隆抗体
可通过使用本领域普通技术人员熟知的方法,用MASP-2多肽或其免疫原性部分免疫动物来制备抗MASP-2的多克隆抗体。参见例如Green等,"Production of Polyclonal Antisera,",载于Immunochemical Protocols(Manson主编),第105页,另详见实施例6。可通过使用佐剂来增加MASP-2多肽的免疫原性,所述佐剂包括无机凝胶(例如氢氧化铝)或弗氏佐剂(完全或不完全)、表面活性剂(例如溶血卵磷脂)、普流罗尼克多元醇(pluronic polyol)、聚阴离子、油乳液、匙孔血蓝蛋白和二硝基苯酚。多克隆抗体一般由马、牛、狗、鸡、大鼠、小鼠、兔、豚鼠、山羊或绵羊等动物产生。或者,用于本发明的抗MASP-2抗体也可得自近似人类的灵长类。在狒狒中产生诊断用和治疗用抗体的通用技术可参见例如Goldenberg等人的国际专利公布号WO 91/11465和Losman,M.J.等,Int.J.Cancer 46:310,1990。然后采用本领域众所周知的标准方法,从这些被免疫过的动物的血液中获得含有免疫活性抗体的血清。
单克隆抗体
在一些实施方案中,MASP-2抑制剂是抗MASP-2单克隆抗体。抗MASP-2单克隆抗体是高度特异性的,针对单一MASP-2表位。本文所用的修饰语“单克隆”是指获自基本同源的抗体群的抗体性质,不得理解为需要通过任何特定方法来产生的抗体。可采用通过培养物中的连续细胞系以提供抗体分子产生的任何技术来获得单克隆抗体,例如Kohler,G.等,Nature256:495,1975中所述的杂交瘤方法,或者可以通过重组DNA方法制备单克隆抗体(参见例如Cabilly的美国专利第4,816,567号)。也可以采用有关技术从噬菌体抗体文库中分离单克隆抗体(参见Clackson,T.等,Nature 352:624-628,1991和Marks,J.D.等,J.Mol.Biol.222:581-597,1991)。这些抗体可以是任何免疫球蛋白类别,包括IgG、IgM、IgE、IgA、IgD及其任何亚类。
例如,可通过将包含MASP-2多肽或其部分的组合物注射给合适的哺乳动物(例如BALB/c小鼠)而获得单克隆抗体。在预定时间之后,从小鼠中取出脾细胞,使之悬浮于细胞培养基中。然后将脾细胞与无限增殖细胞系融合形成杂交瘤。将形成的杂交瘤在细胞培养基中培养,对它们产生抗MASP-2单克隆抗体的能力进行筛选。进一步描述抗MASP-2单克隆抗体产生的实例详见实施例7。(也可参见Current Protocols in Immunology,第1卷,John Wiley&Sons,第2.5.1-2.6.7页,1991)。
可通过使用转基因小鼠来获得人单克隆抗体,所述转基因小鼠已被工程改造以在响应抗原攻击时产生特异性人抗体。在这种技术中,将人免疫球蛋白重链和轻链基因座元件引入得自胚胎干细胞系的小鼠品系中,所述胚胎干细胞系含有被定向破坏的内源免疫球蛋白重链和轻链基因座。该转基因小鼠可合成对人抗原(例如本文所述MASP-2抗原)特异性的人抗体,可使用该小鼠来产生分泌人MASP-2抗体的杂交瘤,其通过采用常规Kohler-Milstein技术,将来自这些动物的B细胞与合适的骨髓瘤细胞系融合(详见实施例7)。具有人免疫球蛋白基因组的转基因小鼠是市售的(例如购自Abgenix,Inc.,Fremont CA和Medarex,Inc.,Annandale,N.J.)。用于从转基因小鼠中获得人抗体的方法描述于例如Green,L.L.等,Nature Genet.7:13,1994;Lonberg,N.等,Nature 368:856,1994和Taylor,L.D.等,Int.Immun.6:579,1994。
可通过各种已确立的技术从杂交瘤培养物中分离和纯化单克隆抗体。这些分离技术包括用A蛋白琼脂糖凝胶亲和层析、大小排阻层析和离子交换层析(参见例如Coligan,第2.7.1-2.7.12页和第2.9.1-2.9.3页、Baines等,"Purification of Immunoglobulin G(IgG)(免疫球蛋白G(IgG)的纯化)",载于Methods in Molecular Biology,The Humana Press,Inc.,第10卷,第79-104页,1992)。
多克隆抗体、单克隆抗体或得自噬菌体的抗体一旦产生,首先便要测定其对MASP-2结合的特异性。可以利用本领域技术人员已知的各种测定法来检测特异性结合MASP-2的抗体。示例性的测定法包括通过标准方法进行的蛋白质印迹法或免疫沉淀分析法(描述于例如Ausubel等)、免疫电泳、酶联免疫吸附测定、斑点印迹法、抑制或竞争测定法以及夹心测定法(描述于Harlow和Land,Antibodies:A Laboratory Manual,Cold Spring Harbor Laboratory Press,1988)。一旦鉴定出特异性结合MASP-2的抗体,便通过以下几种测定法之一,测定抗MASP-2抗体作为MASP-2抑制剂发挥作用的能力:例如凝集素特异性C4裂解测定法(描述于实施例2)、C3b沉积测定法(描述于实施例2)或者C4b沉积测定法(描述于实施例2)。
本领域普通技术人员可以容易地测定抗MASP-2单克隆抗体的亲和力(参见例如Scatchard,A.,NY Acad.Sci.51:660-672,1949)。在一个实施方案中,用于本发明方法的抗MASP-2单克隆抗体能结合MASP-2,其结合亲和力<100nM,优选<10nM,最优选<2nM。
嵌合/人源化抗体
用于本发明方法的单克隆抗体包括嵌合抗体以及这些抗体的片段,其中重链和/或轻链部分与得自特定物种的抗体的相应序列相同或同源,或者属于特定抗体类别或亚类,而链的其余部分与得自另一物种的抗体的相应序列相同或同源,或者属于另一抗体类别或亚类(Cabilly的美国专利第4,816,567号和Morrison,S.L.等,Proc.Nat’l Acad.Sci.USA 81:6851-6855,1984)。
用于本发明的一种嵌合抗体的形式是人源化单克隆抗MASP-2抗体。非人(例如鼠)抗体的人源化形式是嵌合抗体,含有得自非人免疫球蛋白的最小序列。通过将非人(例如小鼠)互补决定区(CDR)从小鼠免疫球蛋白的可变重链和可变轻链转移到人可变区,从而产生人源化单克隆抗体。然后,典型的做法是将人抗体的其余部分代入非人对应部分的构架区。此外,人源化抗体可包括受体抗体或供体抗体中不存在的残基。这些修饰被用来进一步改进抗体性能。一般而言,人源化抗体将包含基本上全部的至少一种、通常两种可变区,其中所有或基本上所有的超变环都对应于非人免疫球蛋白的超变环,所有或基本上所有的Fv构架区都是人免疫球蛋白序列的Fv构架区。人源化抗体任选可包含至少一部分免疫球蛋白恒定区(Fc),通常为人免疫球蛋白的恒定区。更多详情可参见Jones,P.T.等,Nature 321:522-525,1986;Reichmann,L.等,Nature 332:323-329,1988;以及Presta,Curr.Op.Struct.Biol.2:593-596,1992。
用于本发明的人源化抗体包括至少含有MASP-2结合CDR3的人单克隆抗体。此外,可以替换Fc部分以便产生IgA或IgM以及人IgG抗体。这些人源化抗体将具有特殊的临床效用,因为它们特异性地识别人MASP-2,但是却不会引起人体对抗体本身的免疫应答。因此它们更适用于人体的体内给药,尤其是必须重复或长期给药的时候。
由鼠抗MASP-2单克隆抗体获得人源化抗MASP-2抗体的实例见本文实施例10。人源化单克隆抗体的生产技术还记载于例如Jones,P.T.等,Nature 321:522,1986;Carter,P.等,Proc.Nat’l.Acad.Sci.USA 89:4285,1992;Sandhu,J.S.,Crit.Rev.Biotech.12:437,1992;Singer,I.I.等,J.Immun.150:2844,1993;Sudhir(主编),Antibody Engineering Protocols,Humana Press,Inc.,1995;Kelley,"Engineering Therapeutic Antibodies,"载于Protein Engineering:Principles and Practice,Cleland等(主编),John Wiley&Sons,Inc.,第399-434页,1996;以及Queen的美国专利第5,693,762号(1997)。此外,还有从特定鼠抗体区合成人源化抗体的商业实体,例如Protein Design Labs(Mountain View,CA)。
重组抗体
也可使用重组方法制备抗MASP-2抗体。例如,可使用人免疫球蛋白表达文库(可获自例如Stratagene,Corp.,La Jolla,CA)产生人抗体片段(VH、VL、Fv、Fd、Fab或F(ab')2)来制备人抗体。然后使用类似于产生嵌合抗体的技术,将这些片段用以构建完整的人抗体。
抗独特型抗体
一旦鉴定出具有所需抑制活性的抗MASP-2抗体,便可采用本领域众所周知的技术将这些抗体用来产生类似部分MASP-2的抗独特型抗体,参见例如Greenspan,N.S.等,FASEB J.7:437,1993。例如,结合MASP-2并竞争性抑制补体活化所需的MASP-2蛋白的相互作用的抗体可用来产生类似MASP-2蛋白上的MBL结合位点的抗独特型抗体,从而结合并中和MASP-2的结合配体,例如MBL。
免疫球蛋白片段
用于本发明方法的MASP-2抑制剂不仅包括完整的免疫球蛋白分子,而且还包括众所周知的片段,这些片段包括Fab、Fab'、F(ab)2、F(ab')2和Fv片段、scFv片段、双抗体、线性抗体、单链抗体分子以及由抗体片段形成的多特异性抗体。
本领域众所周知的是,只有小部分的抗体分子即互补位参与抗体与其表位的结合(参见例如Clark,W.R.,The Experimental Foundations of Modern Immunology,Wiley&Sons,Inc.,NY,1986)。抗体的pFc'区和Fc区是经典补体途径的效应子,但是不参与抗原结合。其中pFc'区已被酶切割的抗体,或者所产生的没有pFc'区的抗体被称为F(ab')2片段,它保留了完整抗体的抗原结合部位。分离的F(ab')2片段由于有两个抗原结合部位而被称为二价单克隆抗体。类似地,其中Fc区已被酶切割的抗体,或者所产生的没有Fc区的抗体被称为Fab片段,它保留了完整抗体分子的一个抗原结合部位。
抗体片段可通过蛋白水解而获得,例如通过常规方法经胃蛋白酶或木瓜蛋白酶消化完整抗体。例如,可通过用胃蛋白酶进行抗体酶切割来产生抗体片段,从而提供称为F(ab')2的5S片段。该片段可再使用硫醇还原试剂切割,得到3.5S Fab'单价片段。任选可使用二硫键裂解产生的巯基的封端基团来进行裂解反应。作为替代方法,使用胃蛋白酶的酶切割直接产生两个单价Fab片段和一个Fc片段。这些方法记载于例如Goldenberg的美国专利第4,331,647号;Nisonoff,A.等,Arch.Biochem.Biophys.89:230,1960;Porter,R.R.,Biochem.J.73:119,1959;Edelman等,载于Methods in Enzymology,1:422,Academic Press,1967;以及Coligan的第2.8.1-2.8.10页和第2.10.-2.10.4页。
在一些实施方案中,优选使用缺乏Fc区的抗体片段以避免Fc结合Fcγ受体时激活经典补体途径。有几种方法可产生避免与Fcγ受体相互作用的MoAb。例如,单克隆抗体的Fc区可通过使用蛋白水解酶部分消化(例如无花果蛋白酶消化),从而用化学法去除,因此产生例如结合抗原的抗体片段,例如Fab或F(ab)2片段(Mariani,M.等,Mol.Immunol.28:69-71,1991)。或者,可以在构建本文所述的人源化抗体期间使用不结合Fcγ受体的人γ4IgG同种型。也可使用本文所述重组技术来改造缺少Fc结构域的抗体、单链抗体和结合抗原的结构域。
单链抗体片段
或者,可以制备对MASP-2特异性的单一肽链结合分子,其中重链和轻链Fv区相连接。Fv片段可通过肽接头相连,形成单链抗原结合蛋白(scFv)。通过构建包含编码VH和VL结构域的DNA序列的结构基因来制备这些单链抗原结合蛋白,各结构域之间通过寡核苷酸连接。将结构基因插入表达载体中,随后将其引入宿主细胞(例如大肠杆菌)中。重组宿主细胞合成了由接头肽桥接两个V结构域的单一多肽链。scFv的制备方法记载于例如Whitlow等,"Methods:A Companion to Methods in Enzymology"2:97,1991;Bird等,Science 242:423,1988;Ladner的美国专利第4,946,778号;Pack,P.等,Bio/Technology 11:1271,1993。
举例来说,可通过将淋巴细胞体外暴露于MASP-2多肽,并在噬菌体或类似载体中选择抗体展示文库(例如通过使用固定化的或标记的MASP-2蛋白或肽),获得MASP-2特异性scFv。可通过对噬菌体或细菌(如大肠杆菌)上展示的随机肽文库进行筛选而获得编码具有可能的MASP-2多肽结合结构域的多肽的基因。这些随机肽展示文库可用于筛选与MASP-2相互作用的肽。构建和筛选这些随机肽展示文库的技术是本领域众所周知的(Lardner的美国专利第5,223,409号;Lardner的美国专利第4,946,778号;Lardner的美国专利第5,403,484号;Lardner的美国专利第5,571,698号;以及Kay等,Phage Display of Peptides and Proteins,Academic Press,Inc.,1996),且随机肽展示文库和用于筛选这些文库的试剂盒是市售的,例如购自CLONTECH Laboratories,Inc.(Palo Alto,Calif.)、Invitrogen Inc.(San Diego,Calif.)、New England Biolabs,Inc.(Beverly,Mass.)以及Pharmacia LKB Biotechnology Inc.(Piscataway,N.J.)。
用于本发明这个方面的抗MASP-2抗体片段的另一种形式是编码单一互补决定区(CDR)的肽,其结合MASP-2抗原的表位并抑制MASP-2依赖性补体活化。可通过构建编码目标抗体CDR的基因而获得CDR肽(“最小识别单元”)。例如可通过使用聚合酶链式反应从抗体生成细胞的RNA合成可变区,从而制备这些基因(参见例如Larrick等,Methods:A Companion to Methods in Enzymology2:106,1991;Courtenay-Luck,"Genetic Manipulation of Monoclonal Antibodies(单克隆抗体的遗传操作)",载于Monoclonal Antibodies:Production,Engineering and Clinical Application,Ritter等(主编),第166页,Cambridge University Press,1995;以及Ward等,"Genetic Manipulation and Expression of Antibodies(抗体的遗传操作和表达)",载于Monoclonal Antibodies:Principles and Applications,Birch等(主编),第137页,Wiley-Liss,Inc.,1995)。
将本文所述MASP-2抗体给予有需要的受试者以抑制MASP-2依赖性补体活化。在一些实施方案中,MASP-2抑制剂是效应子功能降低的高亲和性人或人源化单克隆抗MASP-2抗体。
肽抑制剂
在本发明这个方面的一些实施方案中,MASP-2抑制剂包括分离的MASP-2肽抑制剂,其包括抑制MASP-2依赖性补体活化系统的分离的天然肽抑制剂和合成肽抑制剂。本文所用术语“分离的MASP-2肽抑制剂”是指通过结合MASP-2、与MASP-2竞争以结合凝集素途径中的其它识别分子(例如MBL、H-纤维胶凝蛋白、M-纤维胶凝蛋白或L-纤维胶凝蛋白)从而抑制MASP-2依赖性补体活化的肽,和/或直接与MASP-2相互作用以抑制MASP-2依赖性补体活化的肽,所述肽是基本纯化的,其基本上没有其它与之一起存在于自然界以达到实用和适于其既定用途程度的物质。
已经使用肽抑制剂成功地在体内阻碍了蛋白间的相互作用和催化位点。例如,最近,结构上与LFA-1有关的粘着分子的肽抑制剂已获准临床用于凝血病(Ohman,E.M.等,European Heart J.16:50-55,1995)。研究揭示短的线性肽(<30个氨基酸)防止或阻碍整联蛋白依赖性粘附(Murayama,O.等,J.Biochem.120:445-51,1996)。还使用长度范围为25-200个氨基酸残基的较长肽,成功阻断了整联蛋白依赖性粘附(Zhang,L.等,J.Biol.Chem.271(47):29953-57,1996)。一般而言,较长的肽抑制剂具有比短肽高的亲和性和/或较慢的解离速度,因此是更有效的抑制剂。研究还表明,环肽抑制剂是有效的整联蛋白体内抑制剂,用于治疗人炎性疾病(Jackson,D.Y.等,J.Med.Chem.40:3359-68,1997)。产生环肽的一种方法包括其中肽的末端氨基酸是半胱氨酸的肽的合成,从而使得肽能够通过末端氨基酸之间的二硫键以环状形式存在,已经证实用于治疗造血系统肿瘤时,改善了亲和性和体内半衰期(例如Larson的美国专利第6,649,592号)。
合成的MASP-2肽抑制剂
通过模拟对于MASP-2功能是重要的靶区的氨基酸序列,来示例性说明用于本发明这个方面方法中的MASP-2抑制肽。用于实施本发明方法的抑制肽的大小范围为约5个氨基酸至约300个氨基酸。表3提供用于实施本发明这个方面的示例性抑制肽的一览表。可通过几种测定法中的一种对候选MASP-2抑制肽作为MASP-2抑制剂起作用的能力进行测定,这些测定法包括例如凝集素特异性的C4裂解测定法(参见实施例2)以及C3b沉积测定法(参见实施例2)。
在一些实施方案中,MASP-2抑制肽得自MASP-2多肽并选自全长的成熟MASP-2蛋白(SEQ ID NO:6),或者得自MASP-2蛋白的特定结构域,例如CUBI结构域(SEQ ID NO:8)、CUBIEGF结构域(SEQ ID N O:9)、EGF结构域(SEQ ID NO:11)以及丝氨酸蛋白酶结构域(SEQ ID NO:12)。如前所述,研究表明CUBEGFCUBII区是二聚化和与MBL结合所需要的(Thielens等,同上)。特别是在鉴定人体带有Asp105至Gly105纯合突变的研究证实,MASP-2的CUBI结构域中的肽序列TFRSDYN(SEQ ID NO:16)参与结合MBL,所述突变导致从MBL复合物上失去MASP-2(Stengaard-Pedersen,K.等,New England J.Med.349:554-560,2003)。
MASP-2抑制肽还可得自MAp19(SEQ ID NO:3)。如实施例30所述,MAp19(SEQ ID NO:3)(亦称sMAP)具有下调被MBL复合物激活的凝集素途径的能力(Iwaki等,J.Immunol.177:8626-8632,2006)。虽然不希望受理论的束缚,但是很有可能的是,sMAP能够占据MBL中的MASP-2/sMAP结合部位,阻止MASP-2与MBL的结合。据报道,sMAP在与纤维胶凝蛋白A缔合时与MASP-2竞争,抑制通过纤维胶凝蛋白A/MASP-2复合物的补体活化(Endo Y.等,Immunogenetics 57:837-844(2005))。
在一些实施方案中,MASP-2抑制肽得自结合MASP-2并参与凝集素补体途径的凝集素蛋白。已经鉴定出参与该途径的几种不同的凝集素,包括甘露聚糖结合凝集素(MBL)、L-纤维胶凝蛋白、M-纤维胶凝蛋白和H-纤维胶凝蛋白(Ikeda,K.等,J.Biol.Chem.262:7451-7454,1987;Matsushita,M.等,J.Exp.Med.176:1497-2284,2000;Matsushita,M.等,J.Immunol.168:3502-3506,2002)。这些凝集素作为同源三聚体亚基的寡聚体而存在于血清中,每个亚基都具有带有糖识别结构域的N端胶原样纤维。已经证实这些不同的凝集素结合MASP-2,凝集素/MASP-2复合物通过裂解蛋白C4和C2而激活补体。H-纤维胶凝蛋白具有24个氨基酸的氨基端区、带有11个Gly-Xaa-Yaa重复的胶原样结构域、12个氨基酸的颈部结构域和207个氨基酸的血纤蛋白原样结构域(Matsushita,M.等,J.Immunol.168:3502-3506,2002)。H-纤维胶凝蛋白结合GlcNAc,并使被得自鼠伤寒沙门氏菌(S.typhimurium)、明尼苏达沙门氏菌(S.minnesota)和大肠杆菌的LPS包被的人红细胞凝集。已经证实H-纤维胶凝蛋白结合MASP-2和MAp19并激活凝集素途径,出处同上。L-纤维胶凝蛋白/P35也结合GlcNAc,已被证实与人血清中的MASP-2和MAp19缔合,并且证实该复合物激活凝集素途径(Matsushita,M.等,J.Immunol.164:2281,2000)。因此,用于本发明的MASP-2抑制肽可包括选自MBL蛋白(SEQ ID NO:21)、H-纤维胶凝蛋白(Genbank检索号NM_173452)、M-纤维胶凝蛋白(Genbank检索号O00602)以及L-纤维胶凝蛋白(Genbank检索号NM_015838)的至少5个氨基酸的区域。
更准确地讲,科学家已经鉴定出MBL上的MASP-2结合部位是在12个Gly-X-Y三联体“GKD GRD GTK GEK GEP GQG LRG LQG POG KLG POG NOG PSG SOG PKG QKG DOG KS”(SEQ ID NO:26)中,该三联体位于MBP胶原样结构域C端部分的铰链和颈之间(Wallis,R.等,J.Biol.Chem.279:14065,2004)。该MASP-2结合部位区在人H-纤维胶凝蛋白和人L-纤维胶凝蛋白中也是高度保守的。研究指出,所有三种包括氨基酸序列“OGK-X-GP”(SEQ ID NO:22)的凝集素蛋白中存在共有结合部位,该氨基酸序列中字母“O”表示羟脯氨酸,字母“X”表示疏水残基(Wallis等,2004,同上)。因此,在一些实施方案中,用于本发明这个方面的MASP-2抑制肽长度为至少6个氨基酸并且包含SEQ ID NO:22。已经证实包括氨基酸序列“GLR GLQ GPO GKL GPO G”(SEQ ID NO:24)并得自MBL的肽体外结合MASP-2(Wallis等,2004,同上)。为增强与MASP-2的结合,可合成在每端邻接两个GPO三联体的肽(“GPO GPO GLR GLQ GPO GKL GPO GGP OGP O”SEQ ID NO:25),以增强天然MBL蛋白中所发现的三股螺旋的形成(详述见Wallis,R.等,J.Biol.Chem.279:14065,2004)。
MASP-2抑制肽也可得自人H-纤维胶凝蛋白,其包括来自H-纤维胶凝蛋白的共有MASP-2结合区的序列“GAO GSO GEK GAO GPQ GPO GPO GKM GPK GEO GDO”(SEQ ID NO:27)。还包括得自人L-纤维胶凝蛋白的肽,其包括来自L-纤维胶凝蛋白的共有MASP-2结合区的序列“GCO GLO GAO GDK GEA GTN GKR GER GPO GPO GKA GPO GPN GAO GEO”(SEQ ID NO:28)。
MASP-2抑制肽还可得自C4裂解部位,例如连接到抗凝血酶III C端部分的C4裂解部位“LQRALEILPNRVTIKANRPFLVFI”(SEQ ID NO:29)(Glove,G.I.等,Mol.Immunol.25:1261(1988))。
表3:示例性的MASP-2抑制肽
注:字母“O”表示羟脯氨酸。字母“X”为疏水残基。
可对得自C4裂解部位的肽以及抑制MASP-2丝氨酸蛋白酶部位的其它肽进行化学修饰,以使其成为不可逆的蛋白酶抑制剂。例如,合适的修饰可包括但是不必限于C端、Asp或Glu、或者添加到功能侧链上的卤甲基酮(Br、Cl、I、F);在氨基或其它功能侧链上的卤乙酰(或其它α-卤乙酰)基团;在氨基端或羧基端或者功能侧链上的环氧基或含亚胺的基团;或者在氨基端或羧基端或者其它功能侧链上的亚氨酸酯。这些修饰提供了通过肽的共价结合而永久抑制酶的这一优势。这可能导致有效剂量较低和/或给予肽抑制剂需要的频率较低。
除了上述抑制肽以外,用于本发明方法的MASP-2抑制肽包括含有按本文所述方法获得的抗MASP-2MoAb的结合MASP-2的CDR3区的肽。用于合成肽的CDR区的序列可以通过本领域已知方法来测定。重链可变区是通常长度范围为100-150个氨基酸的肽。轻链可变区是通常长度范围为80-130个氨基酸的肽。在重链和轻链可变区内部的CDR序列包括大约仅3-25个氨基酸序列,本领域普通技术人员很容易对其进行测序。
本领域技术人员应理解的是,上述的MASP-2抑制肽的基本同源的变异也可具有MASP-2抑制活性。示例性的变异包括但不必限于在题述肽的羧基端或氨基端有插入、缺失、置换和/或添加的氨基酸的肽及其混合物。因此,我们认为这些具有MASP-2抑制活性的同源肽可用于本发明的方法。所述肽还可包括复制基序和具有保守取代的其它修饰。本文其它部分所述的保守变体包括一种氨基酸与具有同样电荷、大小或疏水性等性质的另一种氨基酸的交换。
可以对MASP-2抑制肽进行修饰以增加溶解度和/或使正或负电荷最大化,以便使之更类似于完整蛋白的区段。衍生物可以具有或没有本文所公开肽的精确的一级氨基酸结构,只要衍生物在功能上仍保留所需要的MASP-2抑制特性。所述修饰可包括氨基酸取代,即被通常已知的20种氨基酸之一或另一种氨基酸取代、被附加有所需特征(例如酶降解抗性)的衍生化氨基酸或取代氨基酸取代或者被D氨基酸取代,或者被另外的模拟一种或多种氨基酸或肽的天然构象和功能的分子或化合物(例如糖)取代;氨基酸缺失;氨基酸插入,即插入通常已知的20种氨基酸之一或另一种氨基酸、插入附加有所需特征(例如酶降解抗性)的衍生化氨基酸或取代氨基酸或者插入D氨基酸,或者插入另外的模拟一种或多种氨基酸或肽的天然构象和功能的分子或化合物(例如糖);或者被另外的模拟母体肽的天然构象、电荷分布和功能的分子或化合物(例如糖或核酸单体)取代。肽也可通过乙酰化或酰胺化进行修饰。
可根据肽生物合成、糖生物合成等已知技术合成衍生的抑制肽。开始时,技术人员可根据合适的计算机程序来确定目标肽的构象。一旦知道本文所公开的肽的构象,技术人员便能以合理设计方式来确定能够在一个或多个位点进行什么类别的取代,以便形成的衍生物保留了母体肽的基本构象和电荷分布,但却可以拥有母体肽中不存在的特征或者其特征优于母体肽存在的特征。一旦鉴定出候选衍生分子,就可使用本文所述测定法来测定衍生物,以确定它们是否发挥MASP-2抑制剂的作用。
MASP-2抑制肽的筛选
还可使用分子建模和合理分子设计来产生和筛选模拟MASP-2关键结合区的分子结构并抑制MASP-2的补体活性的肽。用于建模的分子结构包括抗MASP-2单克隆抗体的CDR区,以及已知对MASP-2功能十分重要的目标区域,这些区域包括如前所述的二聚化所需要的区域、涉及结合MBL的区域以及丝氨酸蛋白酶活性部位。用于鉴定结合特定靶的肽的方法是本领域众所周知的。例如,分子印记可用于从头构建大分子结构,例如结合特定分子的肽。参见例如Shea,K.J.,"Molecular Imprinting of Synthetic Network Polymers:The De Novo synthesis of Macromolecular Binding and Catalytic Sites(合成网状聚合物的分子印记:大分子结合部位和催化部位的合成)"TRIP2(5),1994。
举例来说,制备MASP-2结合肽模拟物的一种方法如下。使已知MASP-2结合肽或具有MASP-2抑制作用的抗MASP-2抗体的结合区的功能性单体(模板)聚合。然后去除模板,接着在模板留下的空隙中使第二类单体聚合,得到具有类似于模板的一种或多种所需性质的新分子。除了以这种方式制备肽外,还可制备作为MASP-2抑制剂的其它MASP-2结合分子,例如多糖、核苷、药物、核蛋白、脂蛋白、糖类、糖蛋白、类固醇、脂质和其它生物活性材料。该方法适用于设计各种各样比其天然对应物更稳定的生物学模拟物,因为它们通常是通过功能单体的自由基聚合而成的,产生了具有生物不能降解骨架的化合物。
肽合成
可采用本领域众所周知的技术制备MASP-2抑制肽,例如最初由Merrifield提出的固相合成技术(Merrifield,J.Amer.Chem.Soc.85:2149-2154,1963)。例如,可根据生产商提供的说明书,使用Applied Biosystems 431A肽合成仪(Foster City,Calif.)实现自动合成。其它技术参见例如Bodanszky,M.等,Peptide Synthesis,第二版,John Wiley&Sons,1976,以及本领域技术人员已知的其它参考文献。
还可使用本领域技术人员已知的标准遗传工程技术来制备肽。例如,可通过酶的方法将编码肽的核酸插入表达载体中,进行DNA表达,在所需要的氨基酸存在下将DNA翻译成肽,从而来制备肽。然后,使用层析技术或电泳技术将肽纯化,或者通过将编码肽的序列与编码载体蛋白的核酸序列同步插入表达载体中的载体蛋白方法,所述载体蛋白可以与肽融合,随后再从肽上切下来。可采用层析技术、电泳技术或免疫学技术(例如经载体蛋白的抗体而结合到树脂上)分离蛋白-肽融合物。可使用化学方法或酶方法(例如通过水解酶)来裂解肽。
也可按照常规技术,用重组宿主细胞来生产用于本发明方法的MASP-2抑制肽。为了表达MASP-2抑制肽编码序列,必须将编码肽的核酸分子与控制表达载体转录表达的调节序列有效连接,然后引入宿主细胞中。除了转录调节序列(例如启动子和增强子)之外,表达载体可包括翻译调节序列和标记基因,标记基因适合于选择携带表达载体的细胞。
可用“基因仪”并用亚磷酰胺法等方法来合成编码MASP-2抑制肽的核酸分子。如果例如基因或基因片段合成的应用中需要化学合成的双链DNA,则分别制备每条互补链。短基因(60-80个碱基对)的生产在技术上简单易行,可通过合成互补链,然后将它们退火来实现。对于较长基因的生产来说,要由长度为20-100个核苷酸的单链片段按模块形式装配成合成基因(双链)。有关多核苷酸合成的综述参见例如Glick和Pasternak,"Molecular Biotechnology,Principles and Applications of Recombinant DNA",ASM Press,1994;Itakura,K.等,Annu.Rev.Biochem.53:323,1984;以及Climie,S.等,Proc.Nat’l Acad.Sci.USA87:633,1990。
小分子抑制剂
在一些实施方案中,MASP-2抑制剂是小分子抑制剂,包括具有低分子量的天然和合成物质,例如肽、肽模拟物和非肽抑制剂(包括寡核苷酸和有机化合物)。MASP-2的小分子抑制剂可根据抗MASP-2抗体的可变区的分子结构而产生。
小分子抑制剂也可根据MASP-2的晶体结构采用计算机药物设计来进行设计和产生(Kuntz I.D.等,Science 257:1078,1992)。已经证实大鼠MASP-2的晶体结构(Feinberg,H.等,EMBO J.22:2348-2359,2003)。应用Kuntz等描述的方法,将MASP-2晶体结构坐标输入计算机程序(如DOCK),计算机将输出预期结合MASP-2的小分子结构的清单。这些计算机程序的使用为本领域技术人员所熟知。例如,使用HIV-1蛋白酶抑制剂的晶体结构,通过运用程序DOCK,评价从剑桥晶体学数据库(Cambridge Crystallographic database)中查找到的化合物与酶结合部位的匹配性,来鉴定作为HIV-1蛋白酶抑制剂的独特的非肽配体(Kuntz,I.D.等,J.Mol.Biol.161:269-288,1982;DesJarlais,R.L.等,PNAS 87:6644-6648,1990)。
应用例如实施例7所述的MASP-2结合测定法来筛选用计算机方法鉴定为潜在的MASP-2抑制剂的一系列小分子结构。然后在例如实施例2所述的功能测定法中对被确定为结合MASP-2的小分子进行分析,以确定它们是否抑制MASP-2依赖性补体活化。
MASP-2可溶性受体
一般认为其它合适的MASP-2抑制剂包括MASP-2可溶性受体,可以采用本领域普通技术人员已知技术进行生产。
MASP-2的表达抑制剂
在本发明这个方面的另一实施方案中,MASP-2抑制剂是能够抑制MASP-2依赖性补体活化的MASP-2表达抑制剂。在本发明这个方面的实施中,代表性的MASP-2表达抑制剂包括MASP-2反义核酸分子(例如反义mRNA、反义DNA或反义寡核苷酸)、MASP-2核酶以及MASP-2RNAi分子。
反义RNA和反义DNA分子通过与MASP-2mRNA杂交并阻止MASP-2蛋白的翻译而起到直接阻断MASP-2mRNA翻译的作用。反义核酸分子能以许多不同的方式来构建,只要它能够阻碍MASP-2的表达。例如,可通过使MASP-2cDNA(SEQ ID NO:4)的编码区(或其部分)相对于其正常转录方向进行反转以供其互补序列转录,从而构建反义核酸分子。
反义核酸分子通常与一个或多个靶基因的至少一部分基本相同。然而,核酸并不需要完全相同以抑制表达。较高的同源性一般可用来对较短反义核酸分子的使用予以补偿。最小的百分比同一性通常约65%以上,但是较高的百分比同一性可对内源序列的表达发挥更有效的阻抑作用。通常优选基本为大约80%以上的较大的百分比同一性,尽管通常最优选约95%至完全相同。
反义核酸分子不需要具有与靶基因相同的内含子或外显子模式,靶基因的非编码区段可能在获得靶基因表达的反义抑制方面与编码区段是等效的。至少大约8个左右核苷酸的DNA序列可用作反义核酸分子,尽管优选较长的序列。在本发明中,有用的MASP-2抑制剂的代表性实例是反义MASP-2核酸分子,该分子与由SEQ ID NO:4所示核酸序列组成的MASP-2cDNA的互补序列有至少90%同一性。SEQ ID NO:4所示核酸序列编码由SEQ ID NO:5所示的氨基酸序列组成的MASP-2蛋白。
反义寡核苷酸靶向结合MASP-2mRNA是可用于降低MASP-2蛋白合成水平的另一种机制。例如,多聚半乳糖醛酸酶和蝇蕈碱2型乙酰胆碱受体的合成被针对它们相应mRNA序列的反义寡核苷酸所抑制(Cheng的美国专利第5,739,119号和Shewmaker的美国专利第5,759,829号)。此外,用核蛋白细胞周期蛋白、多药抗药性基因(MDG1)、ICAM-1、E-选择蛋白、STK-1、纹状体GABAA受体和人EGF证实了反义抑制的实例(参见例如Baracchini的美国专利第5,801,154号;Baker的美国专利第5,789,573号;Considine的美国专利第5,718,709号;以及Reubenstein的美国专利第5,610,288号)。
文献记载了使得普通技术人员能够确定哪些寡核苷酸可用于本发明的系统,包括使用RNA酶H切割作为转录物中序列可及性的标志来探测靶mRNA的合适部位(Scherr,M.等,Nucleic Acids Res.26:5079-5085,1998;Lloyd等,Nucleic Acids Res.29:3665-3673,2001)。将与MASP-2转录物某些区域互补的反义寡核苷酸混合物加到表达MASP-2的细胞提取物(例如肝细胞)中,进行杂交以产生易受RNA酶H攻击的部位。该方法可以与计算机辅助序列选择相结合,所述计算机辅助的序列选择能够根据序列形成二聚体、发夹结构或其它二级结构的相对能力来预测用于反义组成的最佳序列选择,所述结构可降低或抑制对宿主细胞靶向mRNA的特异性结合。可使用OLIGO引物分析软件(Rychlik,I.,1997)和BLASTN2.0.5算法软件(Altschul,S.F.等,Nucl.Acids Res.25:3389-3402,1997)来进行这些二级结构分析和靶部位选择考量。针对靶序列的反义化合物的长度优选包括约8个至约50个核苷酸。特别优选包括约9个至约35个左右核苷酸的反义寡核苷酸。本发明的发明人认为,实施本发明基于反义寡核苷酸的方法极优选范围为9个至35个核苷酸(即9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34或35个碱基长度左右的核苷酸)的寡核苷酸组成。极优选的MASP-2 mRNA靶区域是位于或邻近AUG翻译起始密码子的区域,以及与mRNA的5’区基本互补的序列,例如MASP 2基因核苷酸序列(SEQ ID NO:4)的-10和+10区域之间的序列。示例性的MASP-2表达抑制剂见表4。
表4:MASP-2的示例性表达抑制剂
如上所述,本文所用术语“寡核苷酸”是指核糖核酸(RNA)或脱氧核糖核酸(DNA)或其模拟物的寡聚体或多聚体。该术语也包括由天然存在的核苷酸、糖和核苷间(骨架)共价键所组成的那些寡聚核酸碱基(oligonucleobase)以及具有非天然存在的修饰的寡核苷酸。这些修饰使得能够引入某些无法通过天然存在的寡核苷酸提供的所需性质,例如毒性降低、抗核酸酶降解的稳定性提高以及细胞摄取增强。在示例性实施方案中,本发明的反义化合物由于磷酸二酯骨架的修饰延长了反义寡核苷酸的寿命而不同于天然DNA,其中磷酸取代基被硫代磷酸酯置换。同样地,寡核苷酸的一端或两端可被一个或多个间插在核酸链内相邻碱基对之间的吖啶衍生物取代。
反义方案的另一替代是使用“RNA干涉”(RNAi)。双链RNA(dsRNA)可在哺乳动物体内引起基因沉默。RNAi的天然功能和共抑制似乎保护基因组不受可移动遗传元件(例如反转录转座子)以及当其活化时在宿主细胞内产生异常RNA或dsRNA的病毒侵袭(参见例如Jensen,J.等,Nat.Genet.21:209-12,1999)。可通过合成两条能够形成双链RNA分子的RNA链来制备双链RNA分子,每条链的长度约为19-25(例如19-23)个核苷酸。例如,用于本发明方法的dsRNA分子可包括相当于表4所列序列及其互补序列的RNA。优选至少一条RNA链具有1-5个核苷酸的3’突出端。合成的RNA链是在形成双链分子的条件下进行组合的。RNA序列可包含SEQ ID NO:4的至少8个核苷酸的部分,总长度为25个核苷酸以下。特定靶的siRNA序列的设计为本领域普通技术人员所掌握。可获得设计siRNA序列并保证表达有至少70%敲低的商业服务(Qiagen,Valencia,Calif)。
dsRNA可作为药物组合物给予并按已知方法进行,其中核酸被引入所需的靶细胞中。通常使用的基因转移方法包括磷酸钙、DEAE-葡聚糖、电穿孔、显微注射和病毒方法。这些方法在Ausubel等,Current Protocols in Molecular Biology,John Wiley&Sons,Inc.,1993中教导。
也可利用核酶来降低MASP-2的量和/或生物活性,例如靶向MASP-2mRNA的核酶。核酶是催化性RNA分子,能够切割具有与核酶序列完全或部分同源的序列的核酸分子。可以设计核酶转基因,该核酶转基因编码与靶RNA特异性配对并在特定位置切割磷酸二酯骨架的RNA核酶,从而使靶RNA功能性失活。在进行这种切割时,核酶本身并无改变,因此能够再循环并切割其它分子。在反义RNA中包括核酶序列赋予了反义RNA切割RNA的活性,从而增加了反义构建体的活性。
用于实施本发明的核酶通常包括杂交区和催化区,杂交区至少约9个核苷酸,与至少部分靶MASP-2mRNA的核苷酸序列互补,催化区适于切割靶MASP-2mRNA(一般参见EPA 0 321 201号;WO88/04300;Haseloff,J.等,Nature 334:585-591,1988;Fedor,M.J.等,Proc.Natl.Acad.Sci.USA87:1668-1672,1990;Cech,T.R.等,Ann.Rev.Biochem.55:599-629,1986)。
核酶可以掺入核酶序列的RNA寡核苷酸的形式直接靶向细胞,或者作为编码所需要的核酶RNA的表达载体而被引入细胞。可与所述用于反义多核苷酸的大致相同的方式使用核酶。
可通过本领域已知的用于DNA和RNA分子合成的任何方法来制备用于本发明方法的反义RNA和DNA、核酶和RNAi分子。这些方法包括本领域众所周知的用于寡脱氧核糖核苷酸和寡核糖核苷酸的化学合成技术,例如固相亚磷酰胺化学合成。或者,可通过编码反义RNA分子的DNA序列的体外和体内转录来产生RNA分子。这种DNA序列可被掺入各种各样的载体中,载体中插入了合适的RNA聚合酶启动子(例如T7或SP6聚合酶启动子)。或者,可将取决于所用启动子而组成型或诱导型合成反义RNA的反义cDNA构建体稳定地引入细胞系中。
可引入各种众所周知的DNA分子修饰,从而增加稳定性和半衰期。有益的修饰包括但不限于将核糖核苷酸或脱氧核糖核苷酸侧翼序列加到分子的5’端和/或3’端,或者在寡脱氧核糖核苷酸骨架内使用硫代磷酸酯或2’O-甲基而不是磷酸二酯酶键。
V.药物组合物和递药方法
剂量
另一方面,本发明提供用于抑制MASP-2依赖性补体活化的不利作用的组合物,所述组合物包含治疗有效量的MASP-2抑制剂和药学上可接受的载体。可以治疗或改善MASP-2依赖性补体活化相关疾病的治疗有效剂量,给予有需要的受试者MASP-2抑制剂。治疗有效剂量是指MASP-2抑制剂足以导致改善疾病症状的量。
可通过标准药学方法,使用实验动物模型(例如实施例3中所述的表达人MASP-2转基因的鼠MASP-2-/-小鼠模型),来测定MASP-2抑制剂的毒性和治疗功效。使用这些动物模型,可使用标准方法来确定NOAEL(无明显不利作用水平)和MED(最小有效剂量)。NOAEL/MED效应之间的剂量比是治疗比率,用NOAEL/MED之比表示。最优选的是治疗比率或指数高的MASP-2抑制剂。从细胞培养物分析和动物研究中获得的数据可用来制定用于人体的剂量范围。MASP-2抑制剂的剂量优选在循环浓度的范围之内,包括几乎无毒性或没有毒性的最小有效剂量。剂量可在这个范围内变化,这取决于所采用的剂型和所使用的给药途径。
对于任何化合物剂型来说,可使用动物模型来评价治疗有效剂量。例如,可在动物模型中配制达得包括MED在内的循环血浆浓度范围的剂量。也可通过例如高效液相层析法来定量测定血浆中MASP-2抑制剂的水平。
除了毒性研究之外,也可根据有生命的受试者中存在的MASP-2蛋白的量以及MASP-2抑制剂的结合亲和性来估计有效剂量。已经证实正常受试人体血清中存在的MASP-2水平在500ng/ml低水平范围内,可使用MASP-2定量测定法来测定具体受试者的MASP-2水平(描述于Moller-Kristensen M.等,J.Immunol.Methods282:159-167,2003)。
包含MASP-2抑制剂的组合物的给药剂量一般根据受试者年龄、体重、身高、性别、一般疾病状况和病史而变化。举例来说,可在大约0.010-10.0mg/kg、优选0.010-1.0mg/kg、更优选0.010-0.1mg/kg受试者体重的剂量范围内给予MASP-2抑制剂,例如抗MASP-2抗体。在某些实施方案中,所述组合物包含抗MASP-2抗体和MASP-2抑制肽的组合。
可根据本领域技术人员熟知的补体测定法来测定特定受试者的本发明MASP-2抑制剂组合物和方法的治疗功效以及合适的剂量。补体产生多种特定的产物。在最近的十年间,已经研发出灵敏精确的测定法,而且大多数的这类活化产物都是市售的,包括小的活化片段C3a、C4a和C5a和大的活化片段iC3b、C4d、Bb和sC5b-9。大多数的这类测定法都利用了与暴露在片段而不是暴露在其所形成的天然蛋白上的新抗原(new antigen/neoantigen)起反应的单克隆抗体,这使得这些测定法非常简单而专一。大多数都依赖于ELISA技术,尽管有时放射免疫测定法仍用于C3a和C5a。放射免疫测定法测定未经加工的片段及其“脱Arg”片段,这些片段是存在于循环中的主要形式。未经加工的片段和C5a脱Arg通过结合细胞表面受体而被迅速清除掉,因而以极低浓度存在,而C3a脱Arg则不结合细胞,只在血浆中蓄积。测定C3a提供了补体活化灵敏的、不依赖途径的标志。可通过测定Bb片段来评价替代途径活化。检测膜攻击途径活化的液相产物sC5b-9,提供了补体被完全激活的证据。因为凝集素途径和经典途径都产生同样的活化产物C4a和C4d,所以测定这两种片段并不提供有关这两条途径中哪一条途径产生了活化产物的任何信息。
另外的药物
包含MASP-2抑制剂的组合物和方法可任选包括一种或多种另外的治疗药物,它们可增强MASP-2抑制剂的活性,或者以相加或协同的方式提供相关治疗功能。例如,一种或多种MASP-2抑制剂可与一种或多种消炎和/或镇痛药物联用。可对另外药物的含量和选择加以确定以获得所需的治疗结果。合适的消炎和/或镇痛药物包括:5-羟色胺受体拮抗剂;5-羟色胺受体激动剂;组胺受体拮抗剂;缓激肽受体拮抗剂;激肽释放酶抑制剂;速激肽受体拮抗剂,包括神经激肽1和神经激肽2受体亚型拮抗剂;降钙素基因相关肽(CGRP)受体拮抗剂;白介素受体拮抗剂;花生四烯酸代谢物合成途径中有活性的酶的抑制剂,包括磷脂酶抑制剂,包括PLA2同工型抑制剂和PLCγ同工型抑制剂、环加氧酶(COX)抑制剂(它可以是COX-1、COX-2的抑制剂或非选择性COX-1和COX-2的抑制剂)、脂加氧酶抑制剂;前列腺素类受体拮抗剂,包括类二十烷酸EP-1和EP-4受体亚型拮抗剂以及血栓烷受体亚型拮抗剂;白三烯受体拮抗剂,包括白三烯B4受体亚型拮抗剂和白三烯D4受体亚型拮抗剂;阿片样物质受体激动剂,包括μ-阿片样物质、δ-阿片样物质和κ-阿片样物质受体亚型激动剂;嘌呤受体激动剂和拮抗剂,包括P2X受体拮抗剂和P2Y受体激动剂;腺苷三磷酸(ATP)敏感性钾通道开放剂;MAP激酶抑制剂;烟酸乙酰胆碱抑制剂;以及α肾上腺素能受体激动剂(包括α-1、α-2和非选择性的α-1和α-2激动剂)。
当用于抑制或治疗再狭窄时,本发明的MASP-2抑制剂可与一种或多种抗再狭窄药物组合进行同时给药。合适的抗再狭窄药物包括:抗血小板药,包括凝血酶抑制剂和受体拮抗剂、腺苷二磷酸(ADP)受体拮抗剂(亦称嘌呤受体1受体拮抗剂)、血栓烷抑制剂和受体拮抗剂以及血小板膜糖蛋白受体拮抗剂;细胞粘附分子的抑制剂,包括选择蛋白抑制剂和整联蛋白自由基;抗趋化药;白介素受体拮抗剂;以及胞内信号转导抑制剂,包括蛋白激酶C(PKC)抑制剂和蛋白酪氨酸磷酸酶、胞内蛋白酪氨酸激酶抑制剂的调节剂、src同源2(SH2)结构域的抑制剂和钙通道拮抗剂。
本发明的MASP-2抑制剂也可与一种或多种其它的补体抑制剂联用。目前还没有补体抑制剂获准用于人体,然而已经证明一些药物体内阻断补体。这类药物中的许多也是有毒的,或者只是部分抑制剂(Asghar,S.S.,Pharmacol.Rev.36:223-44,1984),这些药物的使用限于用作研究工具。K76COOH和萘莫司他甲磺酸盐是两种在移植动物模型中具有一定疗效的药物(Miyagawa,S.等,Transplant Proc.24:483-484,1992)。研究表明低分子量肝素也有效调节补体活性(Edens,R.E.等,Complement Today第96-120页,Basel:Karger,1993)。我们认为这些小分子抑制剂可用作与本发明MASP-2抑制剂联用的药物。
其它天然存在的补体抑制剂可用于与本发明的MASP-2抑制剂联用。补体的生物学抑制剂包括可溶性补体因子1(sCR1)。这是一种可在人细胞的外膜上找到的天然存在的抑制剂。其它膜抑制剂包括DAF、MCP和CD59。已经对重组形式的体外和体内抗补体活性进行了测定。sCR1表明在异种移植中是有效的,异种移植中补体系统(替代和经典两者)在新移植器官灌注血液几分钟内就引起过度反应排斥综合征(hyperactive rejection syndrome)(Platt J.L.等,Immunol.Today 11:450-6,1990;Marino I.R.等,Transplant Proc.1071-6,1990;Johnstone,P.S.等,Transplantation 54:573-6,1992)。sCR1的使用保护并延长了移植器官的存活时间,表明器官存活机制中的补体途径(Leventhal,J.R.等,Transplantation 55:857-66,1993;Pruitt,S.K.等,Transplantation 57:363-70,1994)。
适于与本发明的组合物联用的其它补体抑制剂还包括例如由Alexion Pharmaceuticals,Inc.New Haven Connecticut正在开发的MoAb,以及抗备解素MoAb。
当用于治疗关节炎(例如骨关节炎和类风湿性关节炎)时,本发明的MASP-2抑制剂可与一种或多种软骨保护药联用,软骨保护药可包括一种或多种软骨合成代谢的促进剂和/或一种或多种软骨分解代谢的抑制剂,适当包括同时给药的合成代谢剂(anabolic agent)和分解代谢抑制剂(catabolic inhibitory agent)两者。合适的促合成代谢的软骨保护药包括白介素(IL)受体激动剂,包括IL-4、IL-10、IL-13、rhIL-4、rhIL-10和rhIL-13以及嵌合IL-4、IL-10或IL-13;转化生长因子β超家族激动剂(包括TGF-β、TGF-β1、TGF-β2、TGF-β3),骨形成蛋白包括BMP-2、BMP-4、BMP-5、BMP-6、BMP-7(OP-1)和OP-2/BMP-8,生长分化因子包括GDF-5、GDF-6和GDF-7,重组TGF-β和BMP,以及嵌合TGF-β和BMP;胰岛素样生长因子,包括IGF-1;以及成纤维细胞生长因子,包括bFGF。合适的抑制分解代谢的软骨保护药包括白介素-1(IL-1)受体拮抗剂(IL-1ra),包括可溶性人IL-1受体(shuIL-1R)、rshuIL-1R、rhIL-1ra、抗IL1抗体、AF11567和AF12198;肿瘤坏死因子(TNF)受体拮抗剂(TNF-α),包括sTNFR1和sTNFRII在内的可溶性受体、重组TNF可溶性受体和嵌合TNF可溶性受体,包括嵌合rhTNFR:Fc、Fc融合可溶性受体和抗TNF抗体;环加氧酶-2(COX-2特异性)抑制剂;包括DuP 697、SC-58451、塞来考昔(celecoxib)、罗非考昔(rofecoxib)、尼美舒利(nimesulide)、双氯芬酸(diclofenac)、美洛昔康(meloxicam)、吡罗昔康(piroxicam)、NS-398、RS-57067、SC-57666、SC-58125、氟舒胺(flosulide)、依托度酸(etodolac)、L-745,337和DFU-T-614;促分裂原活化蛋白激酶(MAPK)抑制剂,包括ERK1、ERK2、SAPK1、SAPK2a、SAPK2b、SAPK2d、SAPK3的抑制剂,包括SB 203580、SB 203580碘、SB202190、SB 242235、SB 220025、RWJ 67657、RWJ 68354、FR 133605、L-167307、PD 98059、PD 169316;核因子κB(NFκB)抑制剂,包括咖啡酸苯乙酯(CAPE)、DM-CAPE、SN-50肽、hymenialdisine和吡咯烷酮二硫代氨基甲酸酯;一氧化氮合酶(NOS)抑制剂,包括NG-一甲基-L-精氨酸、1400W、二苯撑碘、S-甲基异硫脲、S-(氨乙基)异硫脲、L-N6-(1-亚氨基乙基)赖氨酸、1,3-PBITU、2-乙基-2-硫代假脲、氨基胍、Nω-硝基-L-精氨酸、Nω-硝基-L-精氨酸甲酯,基质金属蛋白酶(MMP)的抑制剂,包括MMP-1、MMP-2、MMP-3、MMP-7、MMP-8、MMP-9、MMP-10、MMP-11、MMP-12、MMP-13、MMP-14和MMP-15的抑制剂,包括U-24522、米诺环素(minocycline)、4-Abz-Gly-Pro-D-Leu-D-Ala-NHOH、Ac-Arg-Cys-Gly-Val-Pro-Asp-NH2、重组人TIMP1、重组人TIMP2以及磷阿米酮(phosphoramidon);细胞粘附分子,包括整联蛋白激动剂和拮抗剂,包括αVβ3MoAb LM609和锯鳞蝰素(echistatin);抗趋化剂,包括F-Met-Leu-Phe受体、IL-8受体、MCP-1受体和MIP1-I/RANTES受体;胞内信号转导抑制剂,包括(a)蛋白激酶抑制剂,包括(i)蛋白激酶C(PKC)抑制剂(同工酶)包括抑激酶素C(calphostin C)、G-6203和GF 109203X以及(ii)蛋白酪氨酸激酶抑制剂;(b)胞内蛋白酪氨酸磷酸酶(PTP酶)调节剂;和(c)SH2结构域抑制剂(src同源2结构域)。
对于一些应用来说,将本发明的MASP-2抑制剂与痉挛抑制剂联用可能是有利的。例如,对泌尿生殖系统应用来说,包括至少一种平滑肌痉挛抑制剂和/或至少一种抗炎药可能是有利的,对血管手术来说,包括至少一种血管痉挛抑制剂和/或至少一种抗炎药和/或至少一种抗再狭窄药可能是有益的。痉挛抑制剂的合适实例包括:5-羟色胺2受体亚型拮抗剂;速激肽受体拮抗剂;一氧化氮供体;ATP敏感性钾通道开放剂;钙通道拮抗剂;以及内皮缩血管肽受体拮抗剂。
药物载体和递药载体
一般而言,本发明的MASP-2抑制剂组合物当与任何其它所选的治疗药物联用时,适合于包含在药学上可接受的载体中。载体是无毒的、生物相容的,其选择不得对MASP-2抑制剂(以及与其联用的任何其它治疗药物)的生物活性产生不利的影响。肽的示例性药学上可接受的载体描述于Yamada的美国专利第5,211,657号。用于本发明的抗MASP-2抗体和抑制肽可以配制成固体、半固体、凝胶、液体或气体形式制剂,例如片剂、胶囊剂、粉剂、颗粒剂、软膏剂、溶液剂、栓剂(depositories)、吸入剂和注射剂,供口服、胃肠外或外科手术给药。本发明还包括通过将组合物涂敷在医疗装置上进行局部给药等等。
用于通过注射、输注或冲洗和局部递药的胃肠外递药的合适载体包括蒸馏水、磷酸缓冲生理盐水、标准林格氏液或乳酸盐林格氏液、葡萄糖溶液、Hank氏溶液或丙二醇。此外,无菌不挥发油可用作溶剂或悬浮介质。对于此目的,可采用生物相容性油,包括合成的甘油单酯或甘油二酯。此外,脂肪酸(例如油酸)用于注射剂的制备中。可将载体和药物成分配制成为液体制剂、混悬剂、可聚合或不可聚合的凝胶剂、糊剂或药膏。
载体也可包括递药载体以使递药持续(即延长、延缓或调节),或者增强治疗药物的递送、吸收、稳定性或药代动力学特征。这种递药载体可包括但不限于以下实例:由蛋白质、脂质体、糖类、合成有机化合物、无机化合物、聚合水凝胶、共聚水凝胶和聚合物胶束组成的微粒、微球、纳米球、纳米粒。合适的水凝胶和胶束递送系统包括WO 2004/009664A2中公开的PEO:PHB:PEO共聚物和共聚物/环糊精复合物以及美国专利申请公开号2002/0019369A1中公开的PEO和PEO/环糊精复合物。这些水凝胶可局部注射到既定起效部位,或者皮下或肌内注射以形成缓释贮库。
对于关节内递药,MASP-2抑制剂可被装载于上述可注射的液体或凝胶载体、上述可注射的缓释递药载体、或者透明质酸或透明质酸衍生物中。
对于非肽能药物的口服给药,MASP-2抑制剂可装载于惰性填充剂或稀释剂中,例如蔗糖、玉米淀粉或纤维素。
对于局部给药,MASP-2抑制剂可装载于软膏剂、洗剂、乳膏剂、凝胶剂、滴剂、栓剂、喷雾剂、液体制剂或粉剂中或者透皮贴剂的凝胶或微胶囊递送系统中。
各种经鼻和经肺的递送系统正在研发中,包括气雾吸入器、定量吸入器、干粉吸入器和雾化吸入器,可分别适于递送气雾剂、吸入剂或雾化递药载体中的本发明药物。
对鞘内(IT)或脑室内(ICV)递药,合适的无菌递送系统(例如液体制剂;凝胶剂、混悬剂等)可用来给予本发明的药物。
本发明的组合物还可包括生物相容性赋形剂,例如分散剂或润湿剂、悬浮剂、稀释剂、缓冲剂、渗透促进剂、乳化剂、粘合剂、增稠剂、矫味剂(用于口服给药)。
抗体和肽的药物载体
至于抗MASP-2抗体和抑制肽,更准确地讲,可以按注射剂量的所述化合物的溶液剂或混悬剂经胃肠外给予示例性剂型,所述化合物包含在生理上可接受的稀释剂与药物载体内,药物载体可以是无菌液体,例如水、油、盐水、甘油或乙醇。此外,包含抗MASP-2抗体和抑制肽的组合物中可存在例如润湿剂或乳化剂、表面活性剂、pH缓冲物质等辅助物质。药物组合物的其外组分包括油脂(例如动物、植物或合成来源的油脂),例如大豆油和矿物油。一般而言,二元醇例如丙二醇或聚乙二醇是优选的注射溶液剂的液体载体。
还能以长效注射剂或植入制剂的形式给予抗MASP-2抗体和抑制肽,这些制剂可按允许活性剂缓释或脉冲释放的方式来配制。
表达抑制剂的药学上可接受的载体
至于用于本发明方法的表达抑制剂,更准确地讲是提供包含上述表达抑制剂和药学上可接受的载体或稀释剂的组合物。组合物还可包含胶体分散体系。
包括表达抑制剂的药物组合物可包括但不限于溶液剂、乳剂和含有脂质体的制剂。这些组合物可由各种组分制备,包括但不限于预制液体、自乳化固体和自乳化半固体。这些组合物的制备通常包括将表达抑制剂与一种或多种以下的成分相混合:缓冲剂、抗氧化剂、低分子量多肽、蛋白质、氨基酸、糖类(包括葡萄糖、蔗糖或糊精)、螯合剂(例如EDTA)、谷胱甘肽以及其它稳定剂和赋形剂。中性缓冲盐水或者与非特异性血清白蛋白混合的盐水是合适稀释剂的实例。
在一些实施方案中,组合物可被制备和配制成乳剂,乳剂通常是一种液体以液滴形式分散在另一种液体中的非均相系统(参见Idson,载于Pharmaceutical Dosage Forms,第1卷,Rieger和Banker(主编),Marcek Dekke,Inc.,N.Y.,1988)。用于乳剂的天然存在的乳化剂的实例包括阿拉伯树胶、蜂蜡、羊毛脂、卵磷脂和磷脂。
在一个实施方案中,包括核酸的组合物可配制成微乳剂。本文所用的微乳剂是指水、油和两亲物的系统,它是单一的光学各向同性和热力学稳定的液体溶液(参见Rosoff,载于Pharmaceutical Dosage Forms,第1卷)。本发明的方法也可使用脂质体来将反义寡核苷酸转移和递送到所需要的部位。
用于局部给药的表达抑制剂的药物组合物和制剂可包括透皮贴剂、软膏剂、洗剂、乳膏剂、凝胶剂、滴剂、栓剂、喷雾剂、液体制剂和粉剂。可以使用常规药物载体以及水性基质、粉末基质或油性基质和增稠剂等等。
给药方式
可以多种方式给予包含MASP-2抑制剂的药物组合物,这取决于是局部还是全身性给药方式最适于待治疗的疾病。此外,本文所述的关于体外再灌注方法,可通过将本发明的组合物引入再循环血液或血浆来给予MASP-2抑制剂。而且,本发明的组合物可通过将组合物涂布或掺入可植入的医疗装置上面或里面而递送。
全身性递药
本文所用术语“全身性递药”和“全身性给药”是指包括但不限于口服和胃肠外途径,包括肌内(IM)、皮下、静脉内(IV)、动脉内、吸入、舌下、口腔、局部、经皮、经鼻、直肠、阴道和其它给药途径,它们将所递送的药物有效地分散到预期治疗作用的一个或多个部位。用于本发明的全身性递送的优选途径包括静脉内、肌内、皮下和吸入。应当理解的是,对于用于本发明具体组合物中所选用的药物,确切的全身性给药途径将部分地考虑药物对与特定给药途径相关的代谢转化途径的敏感性加以确定。例如,肽能药物可能最适于通过口服以外的途径给药。
可通过任何合适的方法将MASP-2抑制抗体和多肽递送到有需要的受试者中。递送MASP-2抗体和多肽的方法包括经口服、肺部、胃肠外(例如肌内、腹膜内、静脉内(IV)或皮下注射)、吸入(例如经由微细粉剂型)、经皮、经鼻、阴道、直肠或者舌下给药途径,可将其配制成适于各自给药途径的剂型。
举例来说,可以通过将MASP-2抑制抗体和肽应用到能够吸收多肽的身体膜上,例如鼻膜、胃肠膜和直肠膜,而将其引入活体内。通常将多肽和渗透促进剂一起应用到可吸收膜上(参见例如Lee,V.H.L.,Crit.Rev.Ther.Drug Carrier Sys.5:69,1988;Lee,V.H.L.,J.Controlled Release 13:213,1990;Lee,V.H.L.主编,Peptide and Protein Drug Delivery,Marcel Dekke,New York(1991);DeBoer,A.G.等,J.Controlled Release,13:241,1990)。例如,STDHF是梭链孢酸的合成衍生物,是结构上类似于胆盐的甾类表面活性剂,已被用作经鼻递药的渗透促进剂(Lee,W.A.,Biopharm.22,1990年11/12月)。
可以引入与其它分子(例如脂质)缔合的MASP-2抑制抗体和多肽,以保护多肽不被酶降解。例如,共价结合的聚合物、尤其是聚乙二醇(PEG)已被用来保护某些蛋白质不被体内的酶水解,从而延长半衰期(Fuertges,F.等,J.Controlled Release 11:139,1990)。已经报道了许多用于蛋白质递送的聚合物系统(Bae,Y.H.等,J.Controlled Release 9:271,1989;Hori,R.等,Pharm.Res.6:813,1989;Yamakawa,I.等,J.Pharm.Sci.79:505,1990;Yoshihiro,I.等,J.Controlled Release 10:195,1989;Asano,M.等,J.Controlled Release 9:111,1989;Rosenblatt,J.等,J.Controlled Release 9:195,1989;Makino,K.,J.Controlled Release12:235,1990;Takakura,Y.等,J.Pharm.Sci.78:117,1989;Takakura,Y.等,J.Pharm.Sci.78:219,1989)。
最近,开发出血清稳定性和循环半衰期得到改进的脂质体(参见例如Webb的美国专利第5,741,516号)。而且,对脂质体和脂质体样制备物作为可能的药物载体的各种方法进行了综述(参见例如Szoka的美国专利第5,567,434号;Yagi的美国专利第5,552,157号;Nakamori的美国专利第5,565,213号;Shinkarenko的美国专利第5,738,868号以及Gao的美国专利第5,795,587号)。
对于经皮应用,可将MASP-2抑制抗体和多肽与其它合适的成分(例如载体和/或佐剂)混匀。对这些其它成分的性质没有限制,只是对于其既定给药来说必须是药学上可接受的,并且不能降低组合物中活性成分的活性。合适载体的实例包括含或不含纯化胶原的软膏、乳膏、凝胶或混悬液。MASP-2抑制抗体和多肽也可被浸渍到透皮贴剂、膏药和绷带中,优选液体或半液体形式。
可以在为维持治疗效果所需水平而确定的间隔的周期性基础上,全身性给予本发明的组合物。例如,可按每2-4周或者以更低频率的间隔给予组合物(例如经皮下注射)。给药方案将由医师考虑可能影响药物联用的作用的各种因素来确定。这些因素可包括待治疗疾病的进展程度、患者年龄、性别和体重和其它临床因素。各独立药物成分的剂量将随组合物中所包含的MASP-2抑制剂以及任何递药载体(例如缓释递药载体)的存在和性质而变化。此外,可在考虑给药频率和所递送药物的药代动力学特征的变化后对剂量进行调整。
局部递药
本文所用术语“局部”包括药物在预定的局限性作用部位上或其周围的应用,可包括例如局部递送到皮肤或其它受累组织;眼递药;鞘内(IT)、脑室内(ICV)、关节内、腔内、颅内或肺泡内给药、安置或冲洗。可优选能够给予低剂量的局部给药以避免全身性不利作用,以及更精确地控制递药时间和局部递药部位的活性剂浓度。不论患者之间在新陈代谢、血流等方面的变化,局部给药都在目标部位获得已知浓度。通过直接递药方式还使剂量控制得到改进。
MASP-2抑制剂的局部递送可在用手术方法治疗疾病或病症的外科手术的过程中实现,例如在动脉旁路术、动脉粥样硬化斑切除术、激光手术、超声波手术、球囊血管成形术以及支架安置等手术期间。例如,可将MASP-2抑制剂与球囊血管成形手术结合起来给予受试者。球囊血管成形手术包括将连有已排气球囊的导管插入动脉内。将已排气球囊置于动脉粥样硬化斑块附近,给气囊充气使得斑块向血管壁挤压。结果,球囊表面与血管表面的血管内皮细胞层接触。可以按允许药物在动脉粥样硬化斑块部位释放的方式,将MASP-2抑制剂附着到球囊血管成形术导管上。可按照本领域已知标准方法将药物附着在球囊导管上。例如,可将药物保存在球囊导管的隔室中直到球囊充气,此时药物被释放到局部环境中。或者,可将药物浸渍在球囊表面,从而当球囊充气时,药物就接触到动脉壁的细胞。也可在多孔球囊导管中递送药物,参见例如Flugelman,M.Y.等,Circulation 85:1110-1117,1992。另参见已公开的PCT申请WO 95/23161中对于将治疗蛋白附着到球囊血管成形术导管上的示例性方法。同样地,也可将MASP-2抑制剂包入应用于支架上的凝胶或聚合涂层材料中,或者可将MASP-2抑制剂掺入支架材料中,从而支架在血管安置之后便将MASP-2抑制剂洗脱出来。
用于治疗关节炎和其它肌肉骨骼疾病的MASP-2抑制剂组合物可通过关节内注射来局部递送。这些组合物可适当地包括缓释递药载体。作为其中可能需要局部递药的另一个实例,用于治疗泌尿生殖系统疾病的MASP-2抑制剂组合物可适当地滴入膀胱内或者其它泌尿生殖结构中。
医疗装置上的涂层材料
可将MASP-2抑制剂(例如抗体和抑制肽)固定在可植入或可连接的医疗装置表面(或医疗装置内部)。经修饰的表面通常可在植入动物体后与活组织接触。所谓“可植入或可连接的医疗装置”是指在正常操作该装置(例如支架和可植入递药装置)时,被植入或连接到动物体组织的任何装置。这些可植入或可连接的医疗装置可由诸如以下的材料制成:硝酸纤维素、重氮纤维素、玻璃、聚苯乙烯、聚氯乙烯、聚丙烯、聚乙烯、葡聚糖、琼脂糖凝胶(Sepharose)、琼脂、淀粉、尼龙、不锈钢、钛以及生物可降解的和/或生物相容性聚合物。可通过相连接的蛋白质的生物活性不被破坏的任何技术,来使蛋白质与装置连接,例如通过将蛋白质N端和C端残基的一个或两个连接到装置上。也可在蛋白质内一个或多个位点上进行连接。还可以使用多重连接(既在蛋白质内部也在蛋白质两端)。可对可植入或可连接的医疗装置的表面进行修饰以包括用于供蛋白质固定的官能团(例如羧基、酰胺、氨基、醚、羟基、氰基、次氮基、氨磺酰基(sulfanamido)、乙炔基(acetylinic)、环氧基、硅烷、酐、琥珀酰亚胺基、叠氮基)。偶合化学法包括但不限于与MASP-2抗体或抑制肽上可利用的官能团形成酯、醚、酰胺、叠氮化物和氨磺酰衍生物、氰酸酯和其它键。也可通过将亲和标记物序列加到蛋白质上,从而使MASP-2抗体或抑制片段非共价连接,这些标记物例如GST(D.B.Smith和K.S.Johnson,Gene67:31,1988)、多聚组氨酸(E.Hochuli等,J.Chromatog.411:77,1987)或生物素。这些亲和标记物也可用于蛋白质与装置的可逆连接。
还可将蛋白质共价连接到装置体表面上,例如通过医疗装置表面的共价活化。举例来说,可通过以下任一对反应基团将基质细胞蛋白质(matricellular protein)连接到装置体上(配对基团的一个成员存在于装置体表面,另一个成员则存在于基质细胞蛋白质上):羟基/羧酸,产生酯键;羟基/酸酐,产生酯键;羟基/异氰酸酯,产生氨基甲酸乙酯键。可用射频放电等离子体(RFGD)蚀刻,对不具备有用反应基团的装置体表面进行处理来产生反应基团,以便供基质细胞蛋白质沉积(例如,用氧等离子体处理引入含氧基团;用丙基氨基等离子体处理引入氨基)。
可将包括核酸分子(例如反义核酸)的MASP-2抑制剂、编码肽抑制剂的RNAi或DNA植入连接有装置体的多孔基质中。可用于制造表面层的代表性多孔基质是由腱或皮肤胶原制备的多孔基质,例如可从各种商业来源获得(例如Sigma和Collagen Corporation)的胶原基质,或者胶原基质按照参考文献所述方法制备(Jefferies的美国专利第4,394,370号和Koezuka的美国专利第4,975,527号)。一种胶原材料称为UltraFiberTM,可从Norian Corp.(Mountain View,California)获得。
如果需要,也可采用某些聚合基质,包括丙烯酸酯聚合物和乳酸聚合物,例如参见Urist的美国专利第4,526,909号和Urist的美国专利第4,563,489号。有用聚合物的具体实例为原酸酯、酸酐、丙烯-共延胡索酸酯的聚合物,或者一种或多种α-羟基羧酸单体的聚合物(例如α-羟基乙酸(乙醇酸)和/或α-羟基丙酸(乳酸))。
治疗方案
在预防性应用中,将药物组合物给予易患MASP-2依赖性补体活化相关疾病或有患MASP-2依赖性补体活化相关疾病风险的受试者,给药量足以消除或降低疾病症状发展的风险。在治疗性应用中,以足以缓解或至少部分减少疾病症状的治疗有效量,将药物组合物给予疑似患有或已经患有MASP-2依赖性补体活化相关疾病的受试者。在预防兼治疗方案中,可以给予包含MASP-2抑制剂的组合物若干剂,直到在受试者中获得充分的治疗结果。可通过组合物的单次给予或有限的连续给予,来应用本发明的MASP-2抑制剂组合物以治疗急性疾病,例如再灌注损伤或其它外伤。或者,可在较长一段时间内按定期间隔给予组合物,用于治疗慢性病,例如关节炎或银屑病。
本发明的方法和组合物可用来抑制通常由诊断性和治疗性内科和外科手术引起的炎症和相关过程。为了抑制这些过程,可以在围手术期应用本发明的MASP-2抑制剂组合物。本文所用“围手术期”是指在手术前和/或手术中和/或手术后给予本发明抑制剂组合物,即手术前,手术前和手术中,手术前和手术后,手术前、手术中和手术后,手术中,手术中和手术后,或者手术后。围手术期应用可通过将组合物局部给予手术部位来进行,例如通过注射或者连续或间歇冲洗手术部位,或者通过全身性给药。用于围手术期局部递送MASP-2抑制剂溶液剂的合适方法参见Demopulos的美国专利第6,420,432号和Demopulos的美国专利第6,645,168号。用于局部递送包括MASP-2抑制剂的软骨保护组合物的合适方法参见国际PCT专利申请WO 01/07067 A2。用于定向全身性递送包括MASP-2抑制剂的软骨保护组合物的合适方法参见国际PCT专利申请WO 03/063799 A2。
VI.实施例
下面的实施例仅举例说明目前预期的用于实施本发明的最佳方式,但是不得解释为限制本发明。本文所引用的所有文献都通过引用特别结合到本文中。
实施例1
本实施例描述了缺乏MASP-2(MASP-2-/-)但MAp19充足(MAp19+/+)的小鼠品系的产生。
材料与方法:设计打靶载体pKO-NTKV 1901来破坏编码鼠MASP-2C端的三个外显子,包括编码丝氨酸蛋白酶结构域的外显子,见图4。使用PKO-NTKV 1901转染鼠ES细胞系E14.1a(SV129Ola)。选出新霉素抗性和胸苷激酶敏感的克隆。筛选出600个ES克隆,在其中鉴定出4个不同克隆,通过DNA印迹证实含有预期选择的打靶事件(targeting event)和重组事件,见图4。通过胚胎转移由这4个阳性克隆产生嵌合体。然后将嵌合体在遗传背景C57/BL6下回交以产生转基因雄性鼠。将转基因雄性鼠与雌性鼠杂交产生F1,50%的后代显示出MASP 2基因被破坏的杂合性。将杂合小鼠互交以产生纯合的MASP-2缺陷型后代,按1:2:1的比例分别产生杂合小鼠和野生型小鼠。
结果和表型:发现所产生的纯合MASP-2-/-缺陷型小鼠是可存活和能育的,通过DNA印迹证实缺乏MASP-2,从而确认了正确的打靶事件,经RNA印迹证实缺乏MASP-2mRNA,经蛋白质印迹证实缺乏MASP-2蛋白(数据未显示)。使用时间分辨RT-PCR在LightCycler机上进一步证实了存在MAp19mRNA,缺乏MASP-2mRNA。MASP-2-/-小鼠确实如预期那样继续表达MAp19、MASP-1和MASP-3mRNA及蛋白质(数据未显示)。通过LightCycler分析对MASP-2-/-小鼠中备解素、B因子、D因子、C4、C2和C3的mRNA的存在和丰度进行了评价,发现这些成分与野生型同窝对照中的相同(数据未显示)。来自纯合MASP-2-/-小鼠的血浆完全缺乏凝集素途径介导的补体活化和替代途径补体活化,如实施例2进一步描述的。
在纯的C57BL6背景下产生MASP-2-/-品系:将MASP-2-/-小鼠与纯C57BL6品系回交9代后,将MASP-2-/-品系用作实验动物模型。
实施例2
本实施例证实MASP-2是通过替代途径和凝集素途径进行补体活化所必需的。
方法与材料:
凝集素途径特异性C4裂解测定法:C4裂解测定法记载于Petersen等,J.Immunol.Methods257:107(2001),该测定法测定了由来自金黄色葡萄球菌、结合L-纤维胶凝蛋白的脂磷壁酸(LTA)所产生的凝集素途径活化。对实施例11中所述测定法进行了改进,通过用LPS和甘露聚糖或酵母聚糖包被板后,加入得自MASP-2-/-小鼠的血清,来测定由MBL引起的凝集素途径活化,如下文所述。还对该测定法进行了改进以消除由经典途径所致的C4裂解的可能性。这通过使用含有1M NaCl的样品稀释缓冲液来实现,这使得凝集素途径识别组分以高亲和力结合它们的配体,却又阻止了内源C4的活化,从而通过使C1复合物解离而排除了经典途径的参与。简单地说,在改进的测定法中,将血清样品(稀释于高盐(1M NaCl)缓冲液中)加入到配体包被板上,接着加入缓冲液(具有生理浓度的盐)中的恒量纯C4。含有MASP-2的结合识别复合物裂解C4而引起C4b沉积。
测定方法:
1)用稀释于包被缓冲液(15mM Na2CO3,35mM NaHCO3,pH 9.6)的1μg/ml甘露聚糖(M7504Sigma)或任何其它配体(例如下列配体),来包被Nunc Maxisorb微量滴定板(Maxisorb,Nunc,目录号442404,Fisher Scientific)。
下列试剂用于本测定法中:
a.甘露聚糖(1μg/孔甘露聚糖(M7504Sigma),于100μl包被缓冲液中);
b.酵母聚糖(1μg/孔酵母聚糖(Sigma),于100μl包被缓冲液中);
c.LTA(1μg/孔,于100μl包被缓冲液中,或者2μg/孔,于20μl甲醇中);
d.1μg H-纤维胶凝蛋白特异性Mab 4H5,于包被缓冲液中;
e.来自浅绿气球菌(Aerococcus viridans)的PSA(2μg/孔,于100μl包被缓冲液);
f.100μl/孔福尔马林固定的金黄色葡萄球菌DSM20233(OD550=0.5),于包被缓冲液中。
2)将板在4℃下温育过夜。
3)过夜温育后,通过将板与0.1%HSA-TBS封闭缓冲液(0.1%(重量/体积)HSA的10mM Tris-HCl、140mM NaCl、1.5mM NaN3溶液(pH 7.4))温育1-3小时后,用TBS/吐温(tween)/Ca2+(含0.05%吐温20和5mM CaCl2、1mM MgCl2的TBS(pH 7.4))洗板3次,从而使剩余蛋白结合位点饱和。
4)将待测试的血清样品稀释于MBL结合缓冲液(1M NaCl)中,将稀释的样品加到板上,4℃下温育过夜。只加缓冲液的孔用作阴性对照。
5)在4℃下温育过夜之后,将该板用TBS/吐温/Ca2+洗涤3次。然后将人C4(100μl/孔,1μg/ml稀释于BBS(4mM巴比妥,145mM NaCl,2mM CaCl2,1mM MgCl2,pH 7.4)中)加到板上,在37℃下温育90分钟。该板用TBS/吐温/Ca2+再洗涤3次。
6)用碱性磷酸酶缀合的鸡抗人C4c(以1:1000稀释于TBS/吐温/Ca2+中)来检测C4b沉积,将其加到板上,在室温下温育90分钟。然后该板用TBS/吐温/Ca2+洗涤3次。
7)通过加入100μl磷酸对硝基苯酯底物溶液来检测碱性磷酸酶,在室温下温育20分钟,在微量滴定板读板仪上读取OD405。
结果:图6A-B显示来自MASP-2+/+(交叉线)、MASP-2+/-(实心圆)和MASP-2-/-(实心三角)的血清稀释液中在甘露聚糖(图6A)和酵母聚糖(图6B)上C4b沉积的量。图6C显示来自MASP-2-/+小鼠(n=5)和MASP-2-/-小鼠(n=4)的酵母聚糖(白色条形柱)或甘露聚糖(阴影条形柱)包被板上相对于野生型小鼠(n=5)的C4转化酶相对活性,所根据的是测定针对野生型血清标准化的C4b沉积的量。误差棒代表标准差。如图6A-C所示,来自MASP-2-/-小鼠的血浆在甘露聚糖和酵母聚糖包被板上完全缺乏凝集素途径介导的补体活化。这些结果清楚地表明,MASP-2而不是MASP-1或MASP-3为凝集素途径的效应子成分。
C3b沉积测定法:
1)Nunc Maxisorb微量滴定板(Maxisorb,Nunc,目录号442404,Fisher Scientific)用稀释于包被缓冲液(15mM Na2CO3,35mM NaHCO3,H 9.6)的1μg/ml甘露聚糖(M7504 Sigma)或任何其它配体包被,在4℃下温育过夜。
2)该板与0.1%HSA-TBS封闭缓冲液(0.1%(重量/体积)HSA的10mM Tris-HCl,140mM NaCl,1.5mM NaN3溶液,pH 7.4)一起温育1-3小时,从而使剩余的蛋白结合位点饱和。
3)将该板在TBS/吐温/Ca++(含有0.05%吐温20和5mM CaCl2的TBS)中洗涤,将稀释的BBS加到血清样品(4mM巴比妥,145mM NaCl,2mM CaCl2,1mM MgCl2,pH 7.4)中。将只加缓冲液的孔用作阴性对照。在该项测定中,获自野生型或MASP-2-/-小鼠的一组对照血清样品在使用前耗尽C1q。根据供应商的使用说明书,使用兔抗人C1q IgG(Dako,Glostrup,Denmark)包被的偶联蛋白A的Dynabeads(Dynal Biotech,Oslo,Norway)来制备C1q耗尽的小鼠血清。
4)在4℃下温育过夜后,用TBS/吐温/Ca++再洗涤一次,用以1:1000稀释于TBS/吐温/Ca++的多克隆抗人C3c抗体(Dako A 062)来检测被转化和结合的C3。二次抗体是与碱性磷酸酶缀合的山羊抗兔IgG(完整分子)(Sigma Immunochemicals A-3812),按1:10000稀释于TBS/吐温/Ca++。通过加入100μl底物溶液(Sigma快速磷酸对硝基苯酯片套装,Sigma)并在室温下温育,来测定替代补体途径(AP)的存在。通过在微量滴定板读板仪中测定405nm的吸收,来定量监测水解情况。采用血浆/血清样品的连续稀释液来绘制用于各项分析的标准曲线。
结果:图7A和图7B所示结果来自若干小鼠的合并血清。交叉线表示MASP-2+/+血清,实心圆表示C1q耗尽的MASP-2+/+血清,空心方块表示MASP-2-/-血清,空心三角表示C1q耗尽的MASP-2-/-血清。如图7A-B所示,在C3b沉积测定中所测得的MASP-2-/-小鼠的血清在甘露聚糖(图7A)和酵母聚糖(图7B)包被板上产生水平非常低的C3活化。这个结果清楚地表明,为了启动替代补体途径,需要MASP-2以助于从C3产生最初的C3b。这是预料不到的结果,因为普遍接受的观点认为,补体因子C3、B因子、D因子和备解素形成独立的功能性替代途径,其中C3能够经历自发构象变化成为“C3b样”形式,进而产生液相转化酶iC3Bb,并使C3b分子沉积在活化表面(例如酵母聚糖)上。
重组MASP-2重构MASP-2-/-小鼠血清中的凝集素途径依赖性C4活化
为了证实MASP-2缺乏是MASP-2-/-小鼠凝集素途径依赖性C4活化丧失的直接原因,在上述C4裂解测定中测试了将重组MASP-2蛋白加到血清样品中的作用。按照以下实施例5中所述方法,制备了有功能活性的鼠MASP-2和无催化活性的鼠MASP-2A(其中丝氨酸蛋白酶结构域活性部位上的丝氨酸残基被丙氨酸残基取代)重组蛋白并进行了纯化。将得自4只MASP-2-/-小鼠的合并血清与蛋白质浓度递增的重组鼠MASP-2或无活性的重组鼠MASP-2A一起预温育,按上述方法测定C4转化酶活性。
结果:如图8所示,将有功能活性鼠重组MASP-2蛋白(用空心三角表示)加到获自MASP-2-/-小鼠的血清中,以蛋白质浓度依赖方式恢复了凝集素途径依赖性C4活化,而无催化活性的鼠MASP-2A蛋白(用星号表示)没有恢复C4活化。将图8所示结果针对用合并的野生型小鼠血清所观察到的C4活化结果标准化(用点线表示)。
实施例3
本实施例描述了鼠MASP-2-/-、MAp19+/+且表达人MASP-2转基因(鼠MASP-2敲除,人MASP-2敲入)的转基因小鼠品系的产生。
材料与方法:构建包括人MASP 2基因启动子区并编码人MASP-2的小基因(SEQ ID NO:49),称为“mini hMASP-2”,见图5,它包括头3个外显子(外显子1至外显子3),接着是代表随后8个外显子的编码序列的cDNA序列,从而编码由其内源启动子驱动的全长MASP-2蛋白。将mini hMASP-2构建体注射到MASP-2-/-的受精卵中,以便通过转基因表达人MASP-2来代替缺失的鼠MASP 2基因。
实施例4
本实施例描述了从人血清中分离酶原形式的人MASP-2蛋白。
人MASP-2的分离方法:从人血清分离MASP-2的方法记载于Matsushita等,J.Immunol.165:2637-2642,2000。简单来讲,使用10mM咪唑缓冲液(pH 6.0),使人血清通过酵母甘露聚糖-琼脂糖凝胶柱,所述缓冲液含有0.2M NaCl、20mM CaCl2、0.2mM NPGB、20μM p-APMSF和2%甘露醇。使MASP-1和MASP-2酶原与MBL结合成复合物后,用含有0.3M甘露糖的上述缓冲液洗脱。为了从MBL上分离出酶原MASP-1和MASP-2,将含有所述复合物的制备物加到抗MBL-琼脂糖凝胶中,然后用含有20mM EDTA和1M NaCl的咪唑缓冲液洗脱出MASP。最后,通过抗MASP-1-琼脂糖凝胶而使酶原MASP-1和MASP-2相互分离开来,缓冲液与抗MBL-琼脂糖凝胶所用缓冲液相同。从洗脱液中回收MASP-2,而MASP-1则用0.1M甘氨酸缓冲液(pH 2.2)洗脱。
实施例5
本实施例描述了重组全长人、大鼠和小鼠MASP-2、MASP-2衍生多肽以及无催化活性的MASP-2突变形式的重组表达和蛋白质生产。
全长人、小鼠和大鼠MASP-2的表达:
将人MASP-2全长cDNA序列(SEQ ID NO:4)同样亚克隆到哺乳动物表达载体pCI-Neo(Promega)中,在CMV增强子/启动子区控制下驱动真核表达(描述于Kaufman R.J.等,Nucleic Acids Research 19:4485-90,1991;Kaufman,Methods in Enzymology,185:537-66(1991))。将全长小鼠cDNA(SEQ ID NO:50)和大鼠MASP-2cDNA(SEQ ID NO:53)分别亚克隆到pED表达载体中。然后应用标准磷酸钙转染方法(描述于Maniatis等,1989),将MASP-2表达载体转染到贴壁的中国仓鼠卵巢细胞系DXB1中。用这些构建体转染的细胞生长得非常缓慢,表明所编码的蛋白酶有细胞毒性。
在另一种方法中,将含有由其内源启动子驱动的人MASP-2cDNA的小基因构建体(SEQ ID NO:49)瞬时转染到中国仓鼠卵巢细胞(CHO)中。人MASP-2蛋白被分泌到培养基中,如下述进行分离。
全长无催化活性的MASP-2的表达:
基本原理:在识别亚成分MBL或纤维胶凝蛋白(L-纤维胶凝蛋白、H-纤维胶凝蛋白或M-纤维胶凝蛋白)结合它们各自的糖类模式之后,MASP-2通过自催化裂解激活。导致MASP-2活化的自催化裂解经常发生在从血清分离MASP-2的过程中,或者在重组表达后的纯化期间。为获得更稳定的蛋白质制备物用作抗原,通过用丙氨酸残基置换蛋白酶结构域催化三联体中存在的丝氨酸残基,产生无催化活性形式的MASP-2,称为MASP-2A,在大鼠中(SEQ ID NO:55Ser617变成Ala617);在小鼠中(SEQ ID NO:52Ser617变成Ala617);或在人中(SEQ ID NO:3Ser618变成Ala618)。
为产生无催化活性的人和鼠MASP-2A蛋白,使用表5所示寡核苷酸进行定点诱变。为了将丝氨酸密码子变成丙氨酸密码子,设计了表5中的寡核苷酸使编码有酶活性丝氨酸的人和鼠cDNA区退火,寡核苷酸含有错配。例如,采用PCR寡核苷酸SEQ ID NO:56-59结合人MASP-2cDNA(SEQ ID NO:4),以便扩增自起始密码子到有酶活性丝氨酸的区域及自丝氨酸到终止密码子的区域,从而产生完整开放可读形式的含有Ser618到Ala618突变的突变型MASP-2A。PCR产物在琼脂糖凝胶电泳和条带制备后被纯化,使用标准加尾方法产生单腺苷重叠。然后将加腺苷尾的MASP-2A克隆到pGEM-T easy载体中,再转化到大肠杆菌中。
通过将SEQ ID NO:64和SEQ ID NO:65两种寡核苷酸以等摩尔量结合,在100℃下加热2分钟后,缓慢冷却到室温,通过对这两种寡核苷酸的激酶化(kinasing)和退火,来产生无催化活性的大鼠MASP-2A蛋白。所得到的退火片段具有Pst1和Xba1匹配末端,将该片段插入以替换野生型大鼠MASP-2cDNA(SEQ ID NO:53)的Pst1-Xba1片段从而产生大鼠MASP-2A。
5'GAGGTGACGCAGGAGGGGCATTAGTGTTT 3'(SEQ ID NO:64)
5'CTAGAAACACTAATGCCCCTCCTGCGTCACCTCTGCA 3'(SEQ ID NO:65)。
按下述方法,进一步将人、小鼠和大鼠MASP-2A分别亚克隆到哺乳动物表达载体pED或pCI-Neo并转染到中国仓鼠卵巢细胞系DXB1中。
在另一种方法中,应用Chen等人所述方法构建无催化活性形式的MASP-2(Chen等,J.Biol.Chem.,276(28):25894-25902,2001)。简单来讲,将含有全长人MASP-2cDNA的质粒(描述于Thiel等,Nature 386:506,1997)用Xho1和EcoR1消化,将MASP-2cDNA(参见本文的SEQ ID NO:4)克隆到pFastBac1杆状病毒转移载体(Life Technologies,NY)相应的限制位点中。然后通过用天然区域氨基酸610-625取代编码肽区域氨基酸610-625(SEQ ID NO:13)的双链寡核苷酸,将MASP-2丝氨酸蛋白酶活性位点Ser618变成Ala618,从而产生带有无活性蛋白酶结构域的MASP-2全长多肽。
含有得自人MASP-2的多肽区的表达质粒的构建
采用MASP-2信号肽(SEQ ID NO:5的残基1-15)来产生下面的构建体以分泌不同的MASP-2结构域。通过PCR扩增编码MASP-2(SEQ ID NO:6)残基1-121的区域(相当于N端CUB1结构域)来制备表达人MASP-2CUBI结构域(SEQ ID NO:8)的构建体。通过PCR扩增编码MASP-2(SEQ ID NO:6)残基1-166的区域(相当于N端CUBIEGF结构域)来制备表达人MASP-2CUBIEGF结构域(SEQ ID NO:9)的构建体。通过PCR扩增编码MASP-2(SEQ ID NO:6)残基1-293的区域(相当于N端CUBIEGFCUBII结构域)来制备表达人MASP-2CUBIEGFCUBII结构域(SEQ ID NO:10)的构建体。采用VentR聚合酶,pBS-MASP-2作为模板,根据已确立的PCR方法,经PCR扩增上述结构域。有义引物(5'-CGGGATCCATGAGGCTGCTGACCCTC-3'SEQ ID NO:34)的5’引物序列在PCR产物的5’端引入BamHI限制位点(下划线)。下表5中所示的各MASP-2结构域的反义引物被设计用来在各种PCR产物末端的EcoRI位点(下划线)后引入终止密码子(粗体)。DNA片段一旦被扩增,便用BamHI和EcoRI消化,并克隆到pFastBac1载体相应的位点中。所得构建体用限制酶切作图表征,并通过dsDNA测序来证实。
表5:MASP-2PCR引物
MASP-2的重组真核表达与无酶活性的小鼠、大鼠和人MASP-2A的蛋白质生产应用标准磷酸钙转染方法(Maniatis等,1989),将上述MASP-2和MASP-2A表达构建体转染到DXB1细胞中。在无血清培养基中产生MASP-2A,以确保制备物不被其它血清蛋白污染。每隔一天从汇合细胞中收获培养基(共四次)。所有三个物种的重组MASP-2A水平平均约为1.5mg/升培养基。
MASP-2A蛋白纯化:通过亲和层析法在MBP-A琼脂糖柱上使MASP-2A(上述Ser-Ala突变体)纯化。这种方法能够快速纯化而无需使用外部标记。将MASP-2A(100-200ml培养基用等体积的加样缓冲液(50mM Tris-Cl,pH 7.5,含有150mM NaCl和25mM CaCl2)稀释)加到MBP-琼脂糖亲和柱(4ml)上,柱用10ml加样缓冲液预平衡。另用10ml加样缓冲液洗涤后,在含有1.25M NaCl和10mM EDTA的50mM Tris-Cl(pH 7.5)的1ml组分中洗脱蛋白质。用SDS-聚丙烯酰胺凝胶电泳来鉴定含有MASP-2A的馏分。必要时,MASP-2A通过离子交换层析在MonoQ柱(HR 5/5)上进一步纯化。蛋白质用含有50mM NaCl的50mM Tris-Cl(pH 7.5)透析,加到用同一缓冲液平衡的柱上。洗涤后,另用10ml的0.05-1M NaCl梯度洗脱出结合的MASP-2A。
结果:从200ml培养基中获得产量为0.25-0.5mg的MASP-2A蛋白。由于糖基化作用,所以经MALDI-MS测得77.5kDa的分子量比未修饰多肽的计算值(73.5kDa)大。在每个N-糖基化位点连接有聚糖是所测分子量的原因。MASP-2A作为单一条带在SDS-聚丙烯酰胺凝胶上迁移,证明在生物合成期间没有被蛋白水解加工。通过平衡超速离心法测得的加权平均分子量与糖基化多肽同型二聚体的计算值一致。
重组人MASP-2多肽的生产
生产重组MASP-2和MASP-2A衍生多肽的另一种方法描述于Thielens,N.M.等,J.Immunol.166:5068-5077,2001。简单来讲,使草地贪夜蛾(Spodoptera frugiperda)昆虫细胞(获自Novagen,Madison,WI的Ready-Plaque Sf9细胞)在Sf900II无血清培养基(Life Technologies)中生长并维持,该培养基补充了50IU/ml青霉素和50mg/ml链霉素(Life Technologies)。使粉纹夜蛾(Trichoplusia ni)(High Five)昆虫细胞(由Jadwiga Chroboczek,Institut de Biologie Structurale,Grenoble,France提供)在TC100培养基(Life Technologies)中维持,该培养基含有补充了50IU/ml青霉素和50mg/ml链霉素的10%FCS(Dominique Dutscher,Brumath,France)。采用Bac-to-Bac系统(Life Technologies)来产生重组杆状病毒。杆粒(bacmid)DNA采用Qiagen中量制备纯化系统(Qiagen)纯化,并用来按照生产商所述方案,在cellfectin的Sf900II SFM培养基(Life Technologies)中转染Sf9昆虫细胞。4天后收集重组病毒颗粒,经病毒噬斑测定滴定,按照King和Possee所述方法(King和Possee,载于The Baculovirus Expression System:A Laboratory Guide,Chapman and Hall Ltd.,London,第111-114页,1992)进行扩增。
在Sf900II SFM培养基中,用含有MASP-2多肽的重组病毒在28℃下感染粉纹夜蛾(High Five)细胞(1.75x107个细胞/175cm2组织培养瓶)96小时,感染复数为2。经离心收集上清液,加入二异丙基氟磷酸至终浓度1mM。
MASP-2多肽被分泌到培养基中。将培养物上清液对50mM NaCl、1mM CaCl2、50mM盐酸三乙醇胺(pH 8.1)透析,以1.5ml/分钟的速度加到同一缓冲液平衡的Q-琼脂糖凝胶快流速柱(Amersham Pharmacia Biotech)(2.8x12cm)上。在同一缓冲液中,用1.2升的线性梯度(至350mM NaCl)进行洗脱。通过蛋白质印迹分析法鉴定出含有重组MASP-2多肽的馏分,加入(NH4)2SO4至60%(重量/体积)进行沉淀,4℃静置过夜。将沉淀重悬浮于145mM NaCl、1mM CaCl2、50mM盐酸三乙醇胺(pH 7.4)中,加到同一缓冲液平衡的TSK G3000SWG柱(7.5x 600mm)(Tosohaas,Montgomeryville,PA)上。然后将已纯化的多肽在Microsep微量浓缩器(截留分子量=10,000)(Filtron,Karlstein,Germany)中通过超滤浓缩至0.3mg/ml。
实施例6
本实施例描述了抗MASP-2多肽的多克隆抗体的生产方法。
材料与方法:
MASP-2抗原:用以下分离的MASP-2多肽免疫兔子,以生产多克隆抗人MASP-2抗血清:分离自血清的人MASP-2(SEQ ID NO:6),描述于实施例4;重组人MASP-2(SEQ ID NO:6),含有无活性的蛋白酶结构域(SEQ ID NO:13)的MASP-2A,见实施例4-5;表达的重组CUBI(SEQ ID NO:8)、CUBEGFI(SEQ ID NO:9)和CUBEGFCUBII(SEQ ID NO:10),如上文实施例5所述。
多克隆抗体:已用BCG(卡介苗(bacillus Calmette-Guerin vaccine))免疫的六周龄兔,经注射100μg MASP-2多肽进行免疫,MASP-2多肽按100μg/ml溶于无菌盐溶液。每4周进行注射,通过ELISA测定法监测抗体效价,描述于实施例7。收集培养物上清液,用于通过A蛋白亲和层析法进行抗体纯化。
实施例7
本实施例描述了抗大鼠或人MASP-2多肽的鼠单克隆抗体的生产方法。
材料与方法:
将100μg人或大鼠rMASP-2或rMASP-2A多肽(按实施例4或实施例5中所述制备)皮下注射到8-12周龄的雄性A/J小鼠(Harlan,Houston,Tex.),所述多肽溶于200μl磷酸缓冲盐溶液(PBS)(pH 7.4)中的完全弗氏佐剂(Difco Laboratories,Detroit,Mich.)。在两周的时间间隔下,将50μg溶于不完全弗氏佐剂的人或大鼠rMASP-2或rMASP-2A多肽两次皮下注射到小鼠。在第四周,给小鼠注射溶于PBS的50μg人或大鼠rMASP-2或rMASP-2A多肽,4天后进行融合。
对于每次融合,从经过免疫的小鼠脾脏制备单细胞悬液,用于与Sp2/0骨髓瘤细胞融合。将5x108个Sp2/0和5x108个脾细胞在含有50%聚乙二醇(分子量1450)(Kodak,Rochester,N.Y.)和5%二甲亚砜(Sigma Chemical Co.,St.Louis,Mo.)的培养基中进行融合。然后将细胞调节到每200μl的Iscove培养基(Gibco,Grand Island,N.Y.)悬液1.5x105脾细胞的浓度,培养基中补充10%胎牛血清、100单位/ml青霉素、100μg/ml链霉素、0.1mM次黄嘌呤、0.4μM氨基蝶呤和16μM胸苷。将200微升细胞悬液加到大约二十个96孔微量培养板的各孔中。大约10天后,取出培养物上清液,用于在ELISA测定法中筛选纯化因子MASP-2的反应性。
ELISA测定法:通过加入50μl经纯化的50ng/ml hMASP-2或大鼠rMASP-2(或rMASP-2A)在室温下过夜,包被Immulon 2(Dynatech Laboratories,Chantilly,Va.)微量试验板的各孔。用于包被的低浓度MASP-2使得能够选择高亲和性抗体。在轻轻拍打培养板除去包被溶液后,将200μl的BLOTTO(脱脂奶粉)的PBS溶液加到每个孔中达1小时以封闭非特异性位点。一小时之后,各孔用缓冲液PBST(含有0.05%吐温20的PBS)洗涤。从各融合孔中收集50微升的培养物上清液,并与50μl的BLOTTO混合,然后加到微量试验板的各孔中。温育1小时之后,各孔用PBST洗涤。然后通过与辣根过氧化物酶(HRP)缀合的山羊抗小鼠IgG(对Fc特异性)(Jackson ImmunoResearch Laboratories,West Grove,Pa.)反应来检测结合的鼠抗体,并以1:2,000稀释于BLOTTO中。将含有0.1%3,3,5,5-四甲基联苯胺(Sigma,St.Louis,Mo.)和0.0003%过氧化氢(Sigma)的过氧化物酶底物溶液加到各孔中,显色30分钟。每孔加入50μl 2M H2SO4终止反应。用BioTek ELISA读板仪(BioTek Instruments,Winooski,Vt.)读取反应混合物在450nm的光密度(OD)。
MASP-2结合测定:
在上述MASP-2ELISA测定法中测试为阳性的培养物上清液可在结合测定中进行测定,以测定MASP-2抑制剂对MASP-2的结合亲和性。也可使用类似测定法来确定抑制剂是否结合补体系统中的其它抗原。
通过用MASP-2的磷酸缓冲盐溶液(PBS)(pH 7.4)(20ng/100μl/孔,Advanced Research Technology,San Diego,CA)在4℃下过夜,来包被聚苯乙烯微量滴定板的各孔(96孔培养基结合板,Corning Costar,Cambridge,MA)。吸出MASP-2溶液后,各孔用含有1%牛血清白蛋白(BSA;Sigma Chemical)的PBS在室温下封闭2小时。无MASP-2包被的孔用作背景对照。将在封闭溶液中不同浓度的杂交瘤上清液或已纯化的抗MASP-2MoAb等分溶液加到各孔中。在室温下温育2小时之后,各孔用PBS充分漂洗。通过在封闭溶液中加入过氧化物酶缀合的山羊抗小鼠IgG(Sigma Chemical)在室温下温育1小时,来检测结合MASP-2的抗MASP-2MoAb。该板用PBS再次充分漂洗后,加入100μl 3,3',5,5'-四甲基联苯胺(TMB)底物(Kirkegaard and Perry Laboratories,Gaithersburg,MD)。加入100μl的1M磷酸来猝灭TMB反应,将板在微量板读板仪(SPECTRA MAX 250,Molecular Devices,Sunnyvale,CA)中在450nm下读数。
然后测定来自阳性孔的培养物上清液在功能测定法(例如实施例2所述C4裂解测定法)中抑制补体活化的能力。然后经有限稀释克隆阳性孔中的细胞。再在上述ELISA测定法中测定MoAb与hMASP-2的反应性。使选出的杂交瘤在转瓶中生长,收集耗尽培养物的上清液,通过A蛋白亲和层析法纯化抗体。
实施例8
本实施例描述了用作筛选MASP-2抑制剂模型的表达人MASP-2的MASP-2-/-敲除小鼠的产生。
材料与方法:
将实施例1所述的MASP-2-/-小鼠与实施例3所述的表达人MASP-2转基因构建体(人MASP-2敲入)的MASP-2-/-小鼠进行杂交,为鼠MASP-2-/-、鼠MAp19+、人MASP-2+的后代用来鉴定人MASP-2抑制剂。
这些动物模型可用作鉴定MASP-2抑制剂功效的试验底物,MASP-2抑制剂例如人抗MASP-2抗体、MASP-2抑制肽和非肽以及包含MASP-2抑制剂的组合物。例如,将动物模型暴露于已知触发MASP-2依赖性补体活化的化合物或试剂中,以足以引起所暴露动物的疾病症状减轻的时间和浓度给予动物模型MASP-2抑制剂。
此外,鼠MASP-2-/-、MAp19+、人MASP-2+小鼠可用来产生含有一种或多种涉及MASP-2有关疾病的细胞类型的细胞系,它可用作该疾病的细胞培养模型。由转基因动物产生连续细胞系是本领域众所周知的,例如参见Small,J.A.等,Mol.Cell Biol.,5:642-48,1985。
实施例9
本实施例描述了在表达人MASP-2和人免疫球蛋白的MASP-2敲除小鼠中生产抗人MASP-2人抗体的方法。
材料与方法:
按照实施例1所述方法产生MASP-2-/-小鼠。然后按照实施例3中所述方法构建表达人MASP-2的小鼠。将纯合MASP-2-/-和表达人MASP-2的MASP-2-/-小鼠各自与得自胚胎干细胞系的小鼠杂交,该胚胎干细胞系被改造成包括定向破坏内源免疫球蛋白重链和轻链基因座,并且表达至少一个区段的人免疫球蛋白基因座。优选该人免疫球蛋白基因座的区段包括重链和轻链组分的未重排序列。内源免疫球蛋白基因的失活和外源免疫球蛋白基因的引入都可通过定向同源重组来实现。由这种方法产生的转基因哺乳动物能够使免疫球蛋白组分序列进行功能性重排,并表达由人免疫球蛋白基因编码的各种同种型的抗体库,而不表达内源免疫球蛋白基因。具有这些性质的哺乳动物的产生和性质描述于例如Thomson,A.D.,Nature148:1547-1553,1994和Sloane,B.F.,Nature Biotechnology14:826,1996。其中小鼠抗体基因失活并在功能上被人抗体基因置换的小鼠的遗传工程改造品系是市售的(例如可获自Abgenix,Fremont CA的)。所得到的子代小鼠能够产生适用于人体疗法的抗人MASP-2人MoAb。
实施例10
本实施例描述了人源化鼠抗MASP-2抗体和抗体片段的产生和生产。
按照实施例7所述方法,在雄性A/J小鼠中产生鼠抗MASP-2单克隆抗体。然后按照下述方法,将鼠恒定区用其人对应物置换来产生IgG与抗体Fab片段的嵌合体,使鼠抗体人源化以降低它的免疫原性,所述嵌合体用于抑制本发明的人受试者体内的MASP-2依赖性补体活化的不利作用。
1.由鼠杂交瘤细胞克隆抗MASP-2可变区基因
按照生产商的方案(Biotech,Houston,Tex.),使用RNAzol由分泌抗MASP-2MoAb的杂交瘤细胞(按照实施例7中所述方法获得)中分离出总RNA。采用寡聚dT作为引物,由总RNA合成第一cDNA链。使用免疫球蛋白恒定C区衍生的3’引物和得自前导肽或鼠VH或VK基因的第一构架区的简并引物组作为5’引物对来进行PCR。按Chen和Platsucas所述方法(Chen,P.F.,Scand.J.Immunol.35:539-549,1992)进行锚定PCR。对于克隆VK基因,用Not1-MAK1引物(5'-TGCGGCCGCTGTAGGTGCTGTCTTT-3'SEQ ID NO:38)来制备双链cDNA。将退火衔接子AD1(5'-GGAATTCACTCGTTATTCTCG GA-3'SEQ ID NO:39)和AD2(5'-TCCGAGAATAACGAGTG-3'SEQ ID NO:40)连接到双链cDNA的5’端和3’端。经Notl消化除去3’端衔接子。然后将消化产物用作PCR模板,以AD1寡核苷酸作为5’引物,MAK2(5'-CATTGAAAGCTTTGGGGTAGAAGTTGTTC-3'SEQ ID NO:41)作为3’引物。将大约500bp的DNA片段克隆到pUC19中。选出若干克隆进行序列分析以证实克隆序列包括预期的鼠免疫球蛋白恒定区。Not1-MAK1和MAK2寡核苷酸得自VK区,分别为Cκ基因第一碱基对下游的182bp和84bp。选择包括完整VK和前导肽的克隆。
对于克隆VH基因,使用Not1-MAG1引物(5'-CGCGGCCGCAGCTGCTCAGAGTGTAGA-3'SEQ ID NO:42)来制备双链cDNA。将退火衔接子AD1和AD2连接到双链cDNA的5’端和3’端。经Notl消化除去3’端衔接子。将消化产物用作PCR模板,以AD1寡核苷酸和MAG2(5'-CGGTAAGCTTCACTGGCTCAGGGAAATA-3'SEQ ID NO:43)作为引物。将长度为500-600bp的DNA片段克隆到pUC19中。Not1-MAG1和MAG2寡核苷酸得自鼠Cγ.7.1区,分别为鼠Cγ.7.1基因第一碱基对下游的180bp和93bp。选择包括完整VH和前导肽的克隆。
2.嵌合MASP-2IgG与Fab的表达载体的构建
将上述克隆的VH和VK基因用作PCR反应的模板,以便将Kozak共有序列加到5’端,将剪接供体加到核苷酸序列的3’端。对该序列进行分析确定没有PCR误差后,将VH和VK基因分别插入到含有人C.γ1和C.κ的表达载体盒中,得到pSV2neoVH-huCγ1和pSV2neoV-huCγ。使用CsCl梯度纯化的重链和轻链载体的质粒DNA通过电穿孔转染COS细胞。48小时后,通过ELISA测定培养物上清液,证实存在大约200ng/ml的嵌合IgG。收获细胞,制备总RNA。使用寡聚dT作为引物从总RNA合成第一cDNA链。将该cDNA用作PCR模板以产生Fd和κDNA片段。对于Fd基因,使用5'-AAGAAGCTTGCCGCCACCA TGGATTGGCTGTGGAACT-3'(SEQ ID NO:44)作为5’引物以及得自CH1的3’引物(5'-CGGGATCCTCAAACTTTCTTGTCCACCTTGG-3'SEQ ID NO:45)进行PCR。证实DNA序列含有完整的人IgG1的VH和CH1结构域。在用合适的酶消化后,将Fd DNA片段插入表达载体盒pSV2dhfr-TUS的HindIII和BamHI限制位点,得到pSV2dhfrFd。pSV2质粒是市售的,由不同来源的DNA区段组成:pBR322DNA(细线)含有pBR322DNA复制起点(pBR ori)以及内酰胺酶氨苄青霉素抗性基因(Amp);SV40DNA,由较粗影线表示并标记,含有SV40DNA复制起点(SV40ori)、早期启动子(dhfr和neo基因的5’)以及聚腺苷酸化信号(dhfr和neo基因的3’端)。得自SV40的聚腺苷酸化信号(pA)也位于Fd基因的3’端。
对于κ基因,使用5'-AAGAAAGCTTGCCGCCACCATGTTCTCA CTAGCTCT-3'(SEQ ID NO:46)作为5’引物和得自CK衍生的3’引物(5'-CGGGATCCTTCTCCCTCTAACACTCT-3'SEQ ID NO:47)进行PCR。验证DNA序列含有完整的VK和人CK区域。在用合适的限制酶消化后,将κDNA片段插入表达载体盒pSV2neo-TUS的HindIII和BamHI限制位点,得到pSV2neoK。Fd和κ基因的表达均由HCMV衍生的增强子和启动子元件驱动。因为Fd基因不包括涉及到链间二硫键的半胱氨酸氨基酸残基,所以该重组的嵌合Fab就含有非共价连接的重链和轻链。该嵌合Fab被命名为cFab。
为获得带有重链和轻链间二硫键的重组Fab,可以将上述Fd基因延长以包括得自人IgG1铰链区的额外9个氨基酸(EPKSCDKTH SEQ ID NO:48)的编码序列。编码Fd基因3’端的30个氨基酸的BstEII-BamHI DNA区段可被编码延长的Fd的DNA区段置换,产生pSV2dhfrFd/9aa。
3.嵌合的抗MASP-2IgG的表达和纯化
为产生分泌嵌合的抗MASP-2IgG的细胞系,用经纯化的pSV2neoVH-huC.γ1和pSV2neoV-huCκ的质粒DNA通过电穿孔转染NSO细胞。在0.7mg/ml G418存在下选出被转染的细胞。将细胞在250ml转瓶中用含血清的培养基进行培养。
将100ml旋动培养物的培养物上清液加到10ml PROSEP-A柱(Bioprocessing,Inc.,Princeton,N.J.)上。柱用10个柱床体积的PBS洗涤。结合的抗体用50mM柠檬酸缓冲液(pH3.0)洗脱。将等体积的1M Hepes(pH 8.0)加到含有纯化抗体的馏分中,调节pH到7.0。通过Millipore膜超滤(截留分子量:3,000),用PBS进行缓冲液交换来去除残留的盐。通过BCA方法(Pierce)测得经纯化抗体的蛋白质浓度。
4.嵌合的抗MASP-2Fab的表达和纯化
为产生分泌嵌合的抗MASP-2Fab的细胞系,用经纯化的pSV2dhfrFd(或pSV2dhfrFd/9aa)和pSV2neoκ的质粒DNA通过电穿孔转染CHO细胞。在G418和甲氨蝶呤存在下选出被转染的细胞。在浓度递增的甲氨蝶呤中对所选细胞系进行扩增。经有限稀释对细胞进行单细胞亚克隆。然后将高产单细胞亚克隆细胞系在100ml转瓶中用无血清培养基进行培养。
采用抗MASP-2MoAb的小鼠抗独特型MoAb,通过亲和层析法对嵌合的抗MASP-2Fab进行纯化。可用与匙孔血蓝蛋白(KLH)缀合的鼠抗MASP-2MoAb给小鼠免疫,筛选出能够与人MASP-2竞争的特异性MoAb结合,从而来制备抗独特型MASP-2MoAb。至于纯化,将来自产生cFab或cFab/9aa的旋动培养的CHO细胞的100ml上清液加到与抗独特型MASP-2MoAb偶联的亲和柱上。然后该柱用PBS充分洗涤后,结合的Fab用50mM二乙胺(pH 11.5)洗脱出来。按照上述方法通过缓冲液交换除去残留的盐。通过BCA方法(Pierce)测定经纯化的Fab的蛋白质浓度。
可应用实施例2中所述的抑制测定法来测定嵌合MASP-2IgG、cFab和cFAb/9aa抑制MASP-2依赖性补体途径的能力。
实施例11
本实施例描述了用作功能性筛选的体外C4裂解测定法,从而鉴定能够阻断经由L-纤维胶凝蛋白/P35、H-纤维胶凝蛋白、M-纤维胶凝蛋白或甘露聚糖的MASP-2依赖性补体活化的MASP-2抑制剂。
C4裂解测定法:Petersen,S.V.等人描述了C4裂解测定法(Petersen,S.V.等,J.Immunol.Methods 257:107,200),测定了由结合L-纤维胶凝蛋白的金黄色葡萄球菌的脂磷壁酸(LTA)导致的凝集素途径活化。
试剂:福尔马林固定的金黄色葡萄球菌(DSM20233)如下制备:将细菌在胰胨豆胨血液培养基(tryptic soy blood medium)中于37℃培养过夜,用PBS洗涤三次,然后在室温下于PBS/0.5%福尔马林中固定1小时,再用PBS洗涤三次后,重悬浮于包被缓冲液(15mM Na2CO3、35mM NaHCO3,pH 9.6)中。
测定法:Nunc MaxiSorb微量滴定板(Nalgene Nunc International,Rochester,NY)的各孔用以下的成分包被:100μl福尔马林固定的金黄色葡萄球菌DSM20233(OD550=0.5)的包被缓冲液与1μg L-纤维胶凝蛋白的包被缓冲液。温育过夜之后,各孔用0.1%人血清白蛋白(HSA)的TBS溶液(10mM Tris-HCl,140mM NaCl,pH 7.4)封闭,然后用含有0.05%吐温20和5mM CaCl2的TBS溶液(洗涤缓冲液)洗涤。将人血清样品稀释于20mM Tris-HCl、1M NaCl、10mM CaCl2、0.05%Triton X-100、0.1%HSA(pH 7.4)中,这就防止了内源C4的活化,并使C1复合物(由C1q、C1r和C1s组成)解离。将不同浓度的MASP-2抑制剂(包括抗MASP-2MoAb和抑制肽)加到血清样品中。将稀释的样品加到板中,在4℃下温育过夜。24小时后,各板用洗涤缓冲液充分洗涤,然后向每个孔中加入0.1μg溶于100μl 4mM巴比妥、145mM NaCl、2mM CaCl2、1mM MgCl2(pH 7.4)的经纯化的人C4(按照Dodds,A.W.,Methods Enzymol.223:46,1993所述方法获得)。37℃1.5小时后,各板再次经洗涤,使用碱性磷酸酶缀合的鸡抗人C4c(获自Immunsystem,Uppsala,Sweden),检测C4b沉积,并使用比色底物磷酸对硝基苯酯进行测定。
关于甘露聚糖的C4测定:修改上述测定法以测定经由MBL的凝集素途径活化,在加入与各种MASP-2抑制剂混合的血清之前,用LSP和甘露聚糖包被测定板。
关于H-纤维胶凝蛋白(Hakata Ag)的C4测定:修改上述测定法以测定经由H-纤维胶凝蛋白的凝集素途径活化,在加入与各种MASP-2抑制剂混合的血清之前,用LSP和H-纤维胶凝蛋白包被测定板。
实施例12
下面的测定法证明了野生型和MASP-2-/-小鼠中存在经典途径活化。
方法:微量滴定板(Maxisorb,Nunc,目录号442404,Fisher Scientific)用0.1%人血清白蛋白与10mM Tris、140mM NaCl(pH 7.4)在室温下包被1小时,然后用按1:1000稀释于TBS/吐温/Ca2+中的绵羊抗全血清的抗血清(Scottish Antibody Production Unit,Carluke,Scotland)在4℃下温育过夜,从而原位产生免疫复合物。从野生型和MASP-2-/-小鼠中获得血清样品,加到包被板中。制备对照样品,其中从野生型和MASP-2-/-血清样品中耗尽C1q。按照供应商的使用说明书,使用包被了兔抗人C1q IgG(Dako,Glostrup,Denmark)的A蛋白偶联的Dynabead(Dynal Biotech,Oslo,Norway)来制备C1q耗尽的小鼠血清。将各板在37℃下温育90分钟。用按1:1000稀释于TBS/吐温/Ca++中的多克隆抗人C3c抗体(Dako A 062)来检测结合的C3b。二次抗体是山羊抗兔IgG。
结果:图9表示用IgG与野生型血清、MASP-2-/-血清、耗尽C1q的野生型和耗尽C1q的MASP-2-/-血清包被板上C3b的相对沉积水平。这些结果证明,经典途径在MASP-2-/-小鼠品系中完整无缺。
实施例13
下面的测定法用来通过在经典途径被免疫复合物启动的情况下分析MASP-2抑制剂的作用,从而检测MASP-2抑制剂是否阻断了经典途径。
方法:为了检测MASP-2抑制剂对其中经典途径被免疫复合物启动的补体活化情况的影响,在37℃下,将含有90%NHS的50μl样品一式三份在10μg/ml免疫复合物(IC)或PBS存在下温育,在37℃温育的还有含有200nM抗备解素单克隆抗体的平行样品(+/-IC)一式三份。37℃温育两小时后,将13mM EDTA加到所有样品中终止进一步的补体活化,立即将样品冷却到5℃。在按照生产商使用说明书,使用ELISA试剂盒(Quidel,目录号A015和A009)进行补体活化产物(C3a和sC5b-9)分析之前,将样品保存于-70℃。
实施例14
本实施例证实,凝集素依赖性MASP-2补体活化系统在腹主动脉瘤修复术后的缺血/再灌注阶段中被激活。
实验基本原理和设计:进行腹主动脉瘤(AAA)修复的患者受到缺血/再灌注损伤,这主要是由补体活化介导的。我们研究了MASP-2依赖性凝集素途径的补体活化在患者进行AAA修复的缺血/再灌注损伤中的作用。血清中的甘露聚糖结合凝集素(MBL)的消耗被用来测定再灌注期间发生的MASP-2依赖性凝集素途径活化的量。
患者血清样品的分离:在本研究中共包括23名进行选择性肾下AAA修复的患者和8名进行腹部大手术的对照患者。
对于正进行AAA修复的患者,在手术期间四个规定的时间点从每名患者的桡动脉(经动脉管路)抽取身体血样:时间点1:麻醉诱导;时间点2:刚好在主动脉被钳住之前;时间点3:刚好在取出主动脉钳前;时间点4:再灌注期间。
对于进行腹部大手术的对照患者,在麻醉诱导时以及手术开始之后两小时取身体血样。
MBL水平的测定:应用ELISA技术,测定每名患者血浆样品的甘露聚糖结合凝集素(MBL)水平。
结果:该项研究的结果见图10,该图是表示MBL水平(y轴)在各个不同时间点(x轴)的平均百分比变化的曲线图。MBL的起始值为100%,此后相应地下降。如图10所示,AAA患者(n=23)显示血浆MBL水平显著降低,在AAA之后的缺血/再灌注时,平均降低了大约41%。相反,在进行腹部大手术的对照患者(n=8)的血浆样品中只观察到较小的MBL消耗。
给出数据呈现的明确迹象表明,补体系统的MASP-2依赖性凝集素途径在AAA修复之后的缺血/再灌注阶段被激活。MBL水平的降低似乎与缺血/再灌注损伤有关,因为在手术结束之后被钳住的大血管再灌注时,MBL水平快速显著地下降。相反,进行腹部大手术而无重大缺血/再灌注损伤的患者的对照血清只显示MBL血浆水平略微下降。鉴于补体活化在再灌注损伤中所起的明确作用,我们得出的结论是,缺血内皮细胞中MASP-2依赖性凝集素途径的活化是缺血/再灌注损伤病理的主要因素。因此,可预期特异性瞬时阻断或降低MASP-2依赖性凝集素途径的补体活化对改善临床手术和疾病的结果具有显著有益的治疗作用,所述临床手术和疾病包括一过性缺血损伤,例如心肌梗塞、肠梗塞、烧伤、移植和中风。
实施例15
本实施例描述了MASP-2-/-品系用作动物模型以检测可用于治疗类风湿性关节炎的MASP-2抑制剂。
背景与基本原理:鼠关节炎模型:K/BxN T细胞受体(TCR)转基因(tg)小鼠是最近开发出来的炎性关节炎模型(Kouskoff,V.等,Cell87:811-822,1996;Korganow,A.S.等,Immunity 10:451-461,1999;Matsumoto,I.等,Science 286:1732-1735,1999;Maccioni M.等,J.Exp.Med.195(8):1071-1077,2002)。K/BxN小鼠自发产生自身免疫性疾病,具有人RA的大多数临床、组织学和免疫学特征(Ji,H.等,Immunity 16:157-168,2002)。该鼠病是关节特异性的,但是被T细胞然后被B细胞对泛表达抗原葡萄糖-6-磷酸异构酶("GPI")的自反应性启动然后保持。此外,将关节炎K/BxN小鼠的血清(或者经纯化的抗GPI Ig)转移到健康动物中,在几天之内就引起关节炎。研究还表明,当多克隆抗GPI抗体或IgG1同种型的抗GPI单克隆抗体的混合物注射到健康受体中时诱发了关节炎(Maccioni等,2002)。该鼠模型与人RA有关,因为研究还发现RA患者的血清含有抗GPI抗体,而这在正常个体中并不存在。在这个系统中对C5缺陷型小鼠进行了测试,发现它阻断了关节炎的产生(Ji,H.等,2002,同上)。在C3失效小鼠(C3null mice)中也存在对关节炎的强烈抑制,这就意味着替代途径,然而MBP-A失效小鼠的确发展成关节炎。然而在小鼠中,MBP-C的存在可能弥补了MBP-A的损失。
根据本文所述观察结果,MASP-2在凝集素途径和替代途径两者的启动中起着必不可少的作用,K/BxN关节炎模型用来筛选用作治疗RA的治疗药物的MASP-2抑制剂。
方法:在60日龄时,从关节炎K/BxN小鼠中获得血清,合并,并给MASP-2-/-小鼠(按照实施例1所述方法获得)注射(150-200μl,腹膜内);并在第0天和第2天给用或不用MASP-2抑制剂(本文所述MoAb、抑制肽等等)的同窝对照注射。一组正常小鼠在接受血清注射之前还用MASP-2抑制剂预处理两天。另一组小鼠在第0天接受血清注射,接着在第6天接受MASP-2抑制剂注射。评价随时间变化的临床指数,将每只染病爪评为1分,将只有轻微肿胀的爪评为1/2分。还通过测径器测定踝厚度(厚度定义为与第0天测定值之差)。
实施例16
本实施例描述了在人血浆灌注的兔心脏离体模型中抑制补体介导的组织损伤实验。
背景与基本原理:补体系统的活化是异种移植物超急性排斥的原因。以前的研究表明,超急性排斥可能在抗供体抗体不存在时通过替代途径的活化而发生(Johnston,P.S.等,Transplant Proc.23:877-879,1991)。
方法:为测定分离的抗MASP-2抑制剂(例如按实施例7所述方法制备的抗MASP-2抗体)是否能够抑制组织损伤中的补体途径,可以使用离体模型来测试抗MASP-2MoAb和抗体片段,该模型中单独摘出的兔心脏用稀释的人血浆灌注。这个模型以前显示出由于替代补体途径活化而对兔心肌造成损伤(Gralinski,M.R.等,Immunopharmacology 34:79-88,1996)。
实施例17
本实施例描述了测定嗜中性粒细胞活化的实验方法,该方法用来测定根据本发明方法来治疗凝集素依赖性途径相关疾病的MASP-2抑制剂的有效剂量。
方法:用于嗜中性粒细胞弹性蛋白酶的测定方法描述于Gupta-Bansal,R.等,MolecularImmunol.37:191-201,2000。简单来讲,用双位点夹心测定法(two-site sandwich assay)测定弹性蛋白酶与血清α1-抗胰蛋白酶的复合物,该法利用抗弹性蛋白酶和α1-抗胰蛋白酶的抗体。用抗人弹性蛋白酶抗体(The Binding Site,Birmingham,UK)在PBS中的1:500稀释液在4℃下过夜来包被聚苯乙烯微量滴定板。在吸出抗体溶液后,各孔用含有0.4%HSA的PBS室温封闭2小时。将使用或未用MASP-2抑制剂处理的等分(100μl)的血浆样品加到各孔中。在室温下温育2小时之后,各孔用PBS充分漂洗。通过加入1:500稀释的封闭溶液中的过氧化物酶缀合的α1抗胰蛋白酶抗体,使之在室温下温育1小时,来检测结合的弹性蛋白酶-α1抗胰蛋白酶复合物。板用PBS洗涤后,加入100μl等分的TMB底物试样。通过加入100μl磷酸,猝灭TMB反应物,将板在微量板读板仪中于450nm下读数。
实施例18
本实施例描述了用来测定用于治疗心肌缺血/再灌注的MASP-2抑制剂的动物模型。
方法:心肌缺血/再灌注模型记载于Vakeva等,Circulation 97:2259-2267,1998和Jordan等,Circulation 104(12):1413-1418,2001。可对所述模型加以如下改进用于MASP-2-/-和MASP-2+/+小鼠中。简单来讲,将成年雄性小鼠麻醉。将导管插入颈静脉和气管中,调节啮齿动物呼吸机,保持100%氧气通气量,使呼出的CO2保持在3.5%和5%之间。进行左胸廓切开术,在左冠状动脉起点的3-4mm处进行缝合。缺血前5分钟,给予动物MASP-2抑制剂,例如抗MASP-2抗体(例如剂量范围为0.01-10mg/kg)。然后拉紧冠状动脉附近的缝合线,使之开始缺血,保持30分钟,接着进行4小时再灌注。同法制备假手术动物,但不拉紧缝合线。
补体C3沉积的分析:再灌注之后,从左心室中心区获取免疫组织化学样品,固定,在-80℃下冷冻备用。将组织切片与HRP缀合的山羊抗大鼠C3抗体一起温育。在抗MASP-2抑制剂存在下,对组织切片存在的C3染色进行分析,与假手术的对照动物和MASP-2-/-动物相比较,鉴定出减少体内C3沉积的MASP-2抑制剂。
实施例19
本实施例描述了MASP-2-/-品系用作动物模型以测定MASP-2抑制剂保护移植组织不受缺血/再灌注损伤的能力。
背景/基本原理:已知缺血/再灌注损伤发生在移植期间的供体器官中。组织损伤的程度与缺血时间长短有关,是由补体介导的,正如在缺血和通过使用补体抑制剂(例如可溶性受体1型(CR1))的各种模型中所证实的一样(Weisman等,Science 249:146-151,1990;Mulligan等,J.Immunol.148:1479-1486,1992;Pratt等,Am.J.Path.163(4):1457-1465,2003)。用于移植的动物模型记载于Pratt等,Am.J.Path.163(4):1457-1465,可加以改进用于MASP-2-/-小鼠模型和/或用作MASP-2+/+模型系统,其中对MASP-2抑制剂保护移植组织不受缺血/再灌注损伤的能力进行筛选。在移植之前用灌注液冲洗供体肾,这为将抗MASP-2抑制剂引入供体肾中提供了机会。
方法:将MASP-2-/-和/或MASP-2+/+小鼠麻醉。剖开供体左肾,将主动脉在肾动脉的向头侧和向尾侧结扎。将portex管状导管(Portex Ltd,Hythe,UK)插在结扎部位之间,用含有MASP-2抑制剂(例如抗MASP-2单克隆抗体)(剂量范围为0.01-10mg/kg)的5ml Soltran肾灌注液(Baxter Health Care,UK)灌注肾脏至少5分钟的时间。然后进行肾移植,监测小鼠随时间变化的情况。
移植受体分析:按不同时间间隔收获肾移植物,采用抗C3对组织切片进行分析以确定C3沉积的程度。
实施例20
本实施例描述了胶原诱发的关节炎(CIA)动物模型的用途,该模型用于测定用来治疗类风湿性关节炎(RA)的MASP-2抑制剂。
背景与基本原理:胶原诱发的关节炎(CIA)代表用天然II型胶原免疫之后,在啮齿动物和灵长类易感品系中可诱发的自身免疫性多关节炎,被认为是人类风湿性关节炎(RA)的相关模型(参见Courtney等,Nature 283:666(1980);Trenthan等,J.Exp.Med.146:857(1977))。RA和CIA的特征都是关节发炎、血管翳形成以及软骨和骨侵蚀。CIA易感鼠品系DBA/1LacJ是已开发出来的CIA模型,其中小鼠在用牛II型胶原免疫后,产生了临床上的严重关节炎(Wang等,J.Immunol.164:4340-4347(2000))。将C5缺陷型小鼠品系与DBA/1LacJ杂交,发现所得品系对CIA关节炎的发生具有抗性(Wang等,2000,同上)。
根据本文所述观察结果,MASP-2在凝集素途径和替代途径的启动中都起着必不可少的作用,CIA关节炎模型可用来筛选对用作治疗RA的治疗药物是有效的MASP-2抑制剂。
方法:按照实施例1所述方法产生MASP-2-/-小鼠。然后将MASP-2-/-小鼠与得自DBA/1LacJ品系(The Jackson Laboratory)的小鼠杂交。将F1与其后代互交以产生DBA/lLacJ系中的纯合MASP-2-/-。
胶原免疫按上述方法(Wang等,2000)进行。简单来讲,用溶于0.01M乙酸(浓度为4mg/ml)的牛II型胶原(BCII)或小鼠II型胶原(MCII)(获自Elastin Products,Owensville,MO)免疫野生型DBA/1LacJ小鼠和MASP-2-/-DBA/1LacJ小鼠。将200μg CII和100μg分枝杆菌注射到每只小鼠尾基端部真皮内。在21天后再次免疫小鼠,每天检查关节炎的外观。评价每只染病爪有关关节炎严重程度随时间变化的关节炎指数。
在胶原免疫时,全身性或者在一个或多个关节处局部注射MASP-2抑制剂(例如抗MASP-2单克隆抗体)(剂量范围为0.01mg/kg-10mg/kg),从而在野生型DBA/1LacJ CIA小鼠中对MASP-2抑制剂进行筛选,并按照上述方法评价随时间变化的关节炎指数。可容易地在MASP-2-/-、hMASP-+/+敲入DBA/1LacJ CIA小鼠模型中对抗hMASP-2单克隆抗体作为治疗药物作出评价。
实施例21
本实施例描述了(NZB/W)F1动物模型的用途,该模型用于测定用来治疗免疫复合物介导的肾小球性肾炎的MASP-2抑制剂。
背景与基本原理:新西兰黑×新西兰白(NZB/W)的F1小鼠自发地出现自身免疫综合征,与人免疫复合物介导的肾小球性肾炎有显著的相似性。NZB/W F1小鼠总是在12月龄左右时死于肾小球性肾炎。如前所述,已经证实补体活化在免疫复合物介导的肾小球性肾炎的发病机制中起着重要作用。研究进一步证实,在NZB/W F1小鼠模型中给予抗C5MoAb导致肾小球性肾炎病程显著改善(Wang等,Proc.Natl.Acad.Sci.93:8563-8568(1996))。根据本文所述观察结果,MASP-2在凝集素途径和替代途径的启动中都起着必不可少的作用,NZB/W F1动物模型可用来筛选对用作治疗肾小球性肾炎的治疗药物是有效的MASP-2抑制剂。
方法:按照实施例1所述方法产生MASP-2-/-小鼠。然后将MASP-2-/-小鼠分别与得自NZB和NZW品系(The Jackson Laboratory)的小鼠杂交。将F1与其后代互交以在NZB和NZW遗传背景下产生纯合的MASP-2-/-。为了确定MASP-2在该模型肾小球性肾炎发病机制中的作用,对野生型NZB×NZW小鼠或MASP-2-/-NZB×MASP-2-/-NZW小鼠杂交所产生的F1个体中该病的发生进行了比较。可每隔一周从MASP-2+/+和MASP-2-/-F1小鼠中收集尿样,监测尿蛋白水平中抗DNA抗体的存在(描述于Wang等,1996,同上)。还进行了肾脏组织病理学分析以监测肾小球系膜基质沉积的量和肾小球性肾炎的发生。
NZB/W F1动物模型还用于筛选对于用作治疗肾小球性肾炎的治疗药物是有效的MASP-2抑制剂。在18周龄时,用抗MASP-2抑制剂(例如抗MASP-2单克隆抗体)(剂量范围为0.01mg/kg-10mg/kg)按每周一次或两周一次的频率腹膜内注射到野生型NZB/W F1。使用上述肾小球性肾炎的组织病理学和生物化学标志来评价小鼠中疾病的发生并鉴定对治疗该病是有用的MASP-2抑制剂。
实施例22
本实施例描述了管状环路(tubing loop)作为测定MASP-2抑制剂模型的用途,所述抑制剂可用于预防体外循环(ECC)(例如心肺分流术(CPB)环路)所产生的组织损伤。
背景与基本原理:如上所述,在CPB期间经历ECC的患者遭受全身性炎症反应,这部分是由血液暴露在体外环路的人工表面所引起的,但也是由不依赖于表面的因素引起的,象外伤和缺血/再灌注损伤(Butle,J.等,Ann.Thorac.Surg.55:552-9,1993;Edmunds,L.H.,Ann.Thorac.Surg.66(增刊):S12-6,1998;Asimakopoulos,G.,Perfusion 14:269-77,1999)。研究进一步证实,替代补体途径在CPB环路的补体活化中起着主要作用,CPB环路引起血液与CPB环路的人工表面发生相互作用(参见Kirklin等,1983,1986,讨论同上)。因此,根据本文所述观察结果,MASP-2在凝集素和替代途径的启动中都起着必不可少的作用,管状环路模型可用来筛选对于用作预防或治疗体外暴露所引起的炎症反应的治疗药物是有效的MASP-2抑制剂。
方法:利用上述用于心肺分流术环路(参见Gong等,J.Clinical Immunol.16(4):222-229(1996))的经修改的管状环路模型,描述于Gupta-Bansal等,MolecularImmunol.37:191-201(2000)。简单来讲,从健康受试者中收集新鲜血液到7ml真空管(vacutainer tube)(每ml全血含有7单位肝素)中。向类似于CPB手术期间所使用(例如内径2.92mm;外径3.73mm,长度:45cm)的聚乙烯管中加入1ml血液,用短硅管片封闭成环。在研究中包括将装有肝素化血液与10mM EDTA的对照管作为背景对照。将样品和对照管在水浴中于37℃下垂直旋转1小时。温育之后,将血液样品转移到装有EDTA的1.7ml微量离心管中,使EDTA终浓度为20mM。将样品离心,收集血浆。临旋转前,将MASP-2抑制剂(例如抗MASP-2抗体)加到肝素化血液中。然后对血浆样品进行分析以测定C3a和可溶性C5b-9浓度,如Gupta-Bansal等,2000所述。
实施例23
本实施例描述了啮齿动物盲肠结扎和穿刺(CLP)模型系统的用途,该模型用于测定用来治疗脓毒症或由脓毒症引起的疾病的MASP-2抑制剂,所述疾病包括严重脓毒症、败血症性休克、脓毒症引起的急性呼吸窘迫综合征和全身性炎症反应综合征。
背景与基本原理:如上所述,许多研究已经证实补体活化在脓毒症发病机制中具有重要作用(参见Bone,R.C.,Annals.Internal.Med.115:457-469,1991)。CLP啮齿动物模型是模拟人体中脓毒症临床病程的公认模型,被认为是用于人体脓毒症合理的替代模型(参见Ward,P.,Nature Review Immunology第4卷:133-142(2004))。最近的研究表明,用抗C5a抗体治疗CLP动物导致菌血症减少,存活率大为改善(Huber-Lang等,J.of Immunol.169:3223-3231(2002))。因此,根据本文所述观察结果,MASP-2在凝集素和替代途径的启动中都起着必不可少的作用,CLP啮齿动物模型用于筛选对于用作预防或治疗脓毒症或由脓毒症引起的疾病的治疗药物是有效的MASP-2抑制剂。
方法:对上述CLP模型(Huber-Lang等,2004,同上)进行如下修改。将MASP-2-/-和MASP-2+/+动物麻醉。沿腹中线切开2cm,将盲肠在回盲瓣的下面牢牢地扎住,避免肠梗阻。然后用21号针反复刺穿盲肠。用丝线和皮肤钳将腹部切口一层层缝合(Ethicon,Summerville,NJ)。紧随CLP后,给动物注射MASP-2抑制剂(例如抗MASP-2单克隆抗体)(剂量范围0.01mg/kg-10mg/kg)。在MASP-2-/-、hMASP-+/+敲入CLP小鼠模型中,可容易地对抗hMASP-2单克隆抗体作为治疗药物作出评价。然后使用上述测定法(Huber-Lang等,2004,同上),对小鼠血浆中得自补体的过敏毒素和呼吸爆发的水平进行分析。
实施例24
本实施例描述了阻断MASP-2活性的高亲和性抗MASP-2Fab2抗体片段的鉴定。
背景与基本原理:MASP-2是具有许多独立功能结构域的复杂蛋白质,包括:MBL和纤维胶凝蛋白的结合部位、丝氨酸蛋白酶催化部位、蛋白水解底物C2的结合部位、蛋白水解底物C4的结合部位、MASP-2酶原自身活化的MASP-2裂解部位和两个Ca++结合部位。鉴定出与高亲和性MASP-2结合的Fab2抗体片段,在功能测定法中测定所鉴定出的Fab2片段,以确定它们是否能够阻断MASP-2功能活性。
为了阻断MASP-2功能活性,抗体或Fab2抗体片段必须结合并阻碍MASP-2功能活性所需要的MASP-2上的结构表位。因此,许多或所有的高亲和性结合抗MASP-2Fab2不能抑制MASP-2功能活性,除非它们能结合直接参与MASP-2功能活性的MASP-2的结构表位。
测定抑制凝集素途径C3转化酶形成的功能测定法被用来评价抗MASP-2Fab2的“阻断活性”。已知MASP-2在凝集素途径中的主要生理作用是产生凝集素介导的补体途径的下一个功能成分,即凝集素途径C3转化酶。凝集素途径C3转化酶是蛋白酶切割C3成为C3a和C3b的关键酶复合物(C4bC2a)。MASP-2不是凝集素途径C3转化酶(C4bC2a)的结构成分;然而,需要MASP-2功能活性以产生组成凝集素途径C3转化酶的两个蛋白质成分(C4b、C2a)。此外,为了使MASP-2产生凝集素途径C3转化酶,似乎需要所有上述MASP-2的独立功能活性。出于这些原因,认为用于评价抗MASP-2Fab2的“阻断活性”优选的测定法是测定抑制凝集素途径C3转化酶形成的功能测定法。
高亲和性Fab2的产生:应用人可变轻链和重链抗体序列的噬菌体展示文库以及用于鉴定与所选出的目标配体反应的Fab2的自动抗体筛选技术,来产生抗大鼠MASP-2蛋白(SEQ ID NO:55)的高亲和性Fab2。利用已知量的大鼠MASP-2(~1mg,纯度>85%)蛋白进行抗体筛选。利用三轮扩增以选出具有最高亲和性的抗体。挑选约250个不同的表达抗体片段的目标(hits)用于ELISA筛选。随后对高亲和性目标进行测序以确定不同抗体的独特性。
将50个独特的抗MASP-2抗体纯化,将250μg各经纯化的Fab2抗体用于表征MASP-2结合亲和性和补体途径功能测定,详述如下。
用于评价抗MASP-2Fab2抑制(阻断)活性的测定法
1.测定抑制凝集素途径C3转化酶形成的测定法:
背景:凝集素途径C3转化酶是蛋白酶切割C3成为两个有效促炎片段过敏毒素C3a和调理素C3b的酶复合物(C4bC2a)。C3转化酶的形成似乎是在介导炎症方面的凝集素途径中的关键步骤。MASP-2不是凝集素途径C3转化酶(C4bC2a)的结构成分;因此,抗MASP-2抗体(或Fab2)不会直接抑制之前已存在的C3转化酶的活性。然而,为了产生组成凝集素途径C3转化酶的两个蛋白质成分(C4b、C2a),MASP-2丝氨酸蛋白酶活性是必需的。因此,抑制MASP-2功能活性的抗MASP-2Fab2(即阻断性抗MASP-2Fab2)会抑制凝集素途径C3转化酶从头形成。C3含有独特的高反应性硫酯基团作为其结构部分。在该测定法中当C3转化酶切割C3时,C3b上的硫酯基团可与通过酯键或酰胺键固定在塑料孔底部的大分子上的羟基或氨基形成共价键,从而有利于在ELISA测定法中检测出C3b。
酵母甘露聚糖是凝集素途径的已知激活剂。在测定C3转化酶形成的下列方法中,将用甘露聚糖包被的塑料孔与稀释的大鼠血清在37℃下温育30分钟以激活凝集素途径。然后洗涤各孔,并采用标准ELISA方法测定固定在孔中的C3b。在该测定法中所产生的C3b的量直接反映了从头形成的凝集素途径C3转化酶。在该测定法中测定选定浓度的抗MASP-2Fab2抑制C3转化酶形成和随后C3b产生的能力。
方法:将96孔Costar培养基结合板与稀释于50mM碳酸盐缓冲液(pH 9.5)的甘露聚糖以1μg/50μl/孔在5℃下温育过夜。过夜温育后,各孔用200μl PBS洗涤三次。然后各孔用100μl/孔的1%牛血清白蛋白的PBS溶液封闭,在室温下轻轻搅动温育1小时。然后各孔用200μl PBS洗涤三次。在5℃下,将抗MASP-2Fab2样品稀释于含Ca++和Mg++的GVB缓冲液(4.0mM巴比妥,141mM NaCl,1.0mM MgCl2,2.0mM CaCl2,0.1%明胶,pH 7.4)至选定浓度。在5℃下,将0.5%大鼠血清加到上述样品中,将100μl转移到各孔。将板盖上,在37℃水浴中温育30分钟以便补体活化。将板从37℃水浴转移装有冰-水混合物的容器中进行终止反应。各孔依次用200μl PBS-吐温20(0.05%吐温20的PBS溶液)洗涤5次,用200μl PBS洗涤2次。加入100μl/孔按1:10,000稀释的初次抗体(兔抗人C3c,DAKO A0062),该抗体溶于含有2.0mg/ml牛血清白蛋白的PBS中,在室温下轻轻搅动温育1小时。各孔用200μl PBS洗涤5次。加入100μl/孔按1:10,000稀释的二次抗体(过氧化物酶缀合的山羊抗兔IgG,American Qualex A102PU),该抗体溶于含有2.0mg/ml牛血清白蛋白的PBS中,室温下在振荡器中轻轻搅动温育1小时。各孔用200μl PBS洗涤5次。加入100μl/孔过氧化物酶底物TMB(Kirkegaard&Perry Laboratories),在室温下温育10分钟。通过加入100μl/孔1.0M H3PO4终止过氧化物酶反应,测得OD450。
2.测定抑制MASP-2依赖性C4裂解的测定法
背景:MASP-2的丝氨酸蛋白酶活性是高度特异性的,仅鉴定出用于MASP-2的两种蛋白质底物:C2和C4。C4裂解产生C4a和C4b。抗MASP-2Fab2可结合直接参与C4裂解的MASP-2的结构表位(例如C4的MASP-2结合部位;MASP-2丝氨酸蛋白酶催化部位),从而抑制MASP-2的C4裂解功能活性。
酵母甘露聚糖是凝集素途径的已知激活剂。在测定MASP-2的C4裂解活性的下列方法中,将用甘露聚糖包被的塑料孔与稀释的大鼠血清在37℃下温育30分钟以激活凝集素途径。因为用于该ELISA测定法中的初次抗体仅识别人C4,所以稀释的大鼠血清还补充了人C4(1.0μg/ml)。然后洗涤各孔,采用标准ELISA方法测定固定在孔中的人C4b。在该测定法中,所产生的C4b的量可衡量依赖MASP-2的C4裂解活性。在该测定法中,测定选定浓度的抗MASP-2Fab2抑制C4裂解的能力。
方法:将96孔Costar培养基结合板与稀释于50mM碳酸盐缓冲液(pH 9.5)的甘露聚糖按1μg/50μl/孔在5℃下温育过夜。各孔用200μl PBS洗涤3次。然后各孔用100μl/孔1%牛血清白蛋白的PBS溶液封闭,在室温下轻轻搅动温育1小时。各孔用200μl PBS洗涤3次。在5℃下,将抗MASP-2Fab2样品稀释于含Ca++和Mg++的GVB缓冲液(4.0mM巴比妥,141mM NaCl,1.0mM MgCl2,2.0mM CaCl2,0.1%明胶,pH 7.4)至选定浓度。1.0μg/ml人C4(Quidel)也包括在这些样品中。在5℃下,将0.5%大鼠血清加到上述样品中,将100μl转移到各孔。将板盖上,在37℃水浴中温育30分钟以便补体活化。将板从37℃水浴转移装有冰-水混合物的容器中进行终止反应。各孔用200μl PBS-吐温20(0.05%吐温20的PBS溶液)洗涤5次,然后各孔用200μl PBS洗涤2次。加入100μl/孔以1:700稀释的生物素缀合的鸡抗人C4c(Immunsystem AB,Uppsala,Sweden)的PBS溶液(含有2.0mg/ml牛血清白蛋白(BSA)),在室温下轻轻搅动温育1小时。各孔用200μl PBS洗涤5次。加入100μl/孔的0.1μg/ml过氧化物酶缀合的链霉抗生物素(Pierce Chemical#21126)的PBS溶液(含有2.0mg/ml BSA),在室温下在振荡器中轻轻搅动温育1小时。各孔用200μl PBS洗涤5次。加入100μl/孔过氧化物酶底物TMB(Kirkegaard&Perry Laboratories),在室温下温育16分钟。通过加入100μl/孔1.0M H3PO4终止过氧化物酶反应,测得OD450。
3.抗“天然”大鼠MASP-2的抗大鼠MASP-2Fab2的结合测定法
背景:MASP-2通常作为还包括特异性凝集素分子(甘露糖结合蛋白(MBL)和纤维胶凝蛋白)的MASP-2二聚体复合物存在于血浆中。因此,如果有兴趣研究抗MASP-2Fab2与生理相关形式的MASP-2结合,则重要的是开发出结合测定法,其中利用的是Fab2与血浆“天然”MASP-2之间,而不是与纯化的重组MASP-2之间的相互作用。在该结合测定法中,先将得自10%大鼠血清的“天然”MASP-2-MBL复合物固定在甘露聚糖包被的孔内。然后采用标准ELISA方法,对各种抗固定化“天然”MASP-2的抗MASP-2Fab2的结合亲和性进行了研究。
方法:将96孔Costar高结合板与稀释于50mM碳酸盐缓冲液(pH 9.5)的甘露聚糖按1μg/50μl/孔在5℃下温育过夜。各孔用200μl PBS洗涤3次。各孔用100μl/孔的0.5%脱脂奶粉的PBST溶液(PBS与0.05%吐温20)封闭,在室温下轻轻搅动温育1小时。各孔用200μl TBS/吐温/Ca++洗涤缓冲液(Tris缓冲盐溶液,0.05%吐温20,含有5.0mMCaCl2,pH 7.4)洗涤3次。在冰上制备10%大鼠血清的高盐结合缓冲液(20mM Tris,1.0MNaCl,10mM CaCl2,0.05%Triton-X100,0.1%(重量/体积)牛血清白蛋白,pH 7.4)。每孔加入100μl,在5℃下温育过夜。各孔用200μl TBS/吐温/Ca++洗涤缓冲液洗涤3次。然后各孔用200μl PBS洗涤2次。加入100μl/孔稀释于含有Ca++和Mg++的GVB缓冲液(4.0mM巴比妥,141mM NaCl,1.0mM MgCl2,2.0mM CaCl2,0.1%明胶,pH 7.4)的选定浓度的抗MASP-2Fab2,在室温下轻轻搅动温育1小时。各孔用200μl PBS洗涤5次。加入100μl/孔按1:5000稀释的溶于2.0mg/ml牛血清白蛋白/PBS的HRP缀合的山羊抗Fab2(Biogenesis目录号0500-0099),在室温下轻轻搅动温育1小时。各孔用200μl PBS洗涤5次。加入100μl/孔过氧化物酶底物TMB(Kirkegaard&Perry Laboratories),在室温下温育70分钟。通过加入100μl/孔的1.0M H3PO4终止过氧化物酶反应,测得OD450。
结果:
挑选了约250个不同的与抗大鼠MASP-2蛋白进行高亲和性反应的Fab2用于ELISA筛选。对这些高亲和性Fab2进行了测序以确定不同抗体的独特性,对50个独特的抗MASP-2抗体进行纯化用于进一步分析。250μg各经纯化的Fab2抗体用来表征MASP-2结合亲和性及测定补体途径功能。该分析的结果见表6。
表6:阻断凝集素途径补体活化的抗MASP-2FAB2
如上表6所示,50个所测的抗MASP-2Fab2中,17个Fab2被鉴定为MASP-2阻断性Fab2,有效抑制C3转化酶形成,IC50≤10nM Fab2(34%阳性选中率)。所鉴定出的17个Fab2中,有8个的IC50范围为纳摩尔以下。此外,表6中所示的所有17个MASP-2阻断性Fab2在凝集素途径C3转化酶测定法中基本上完全抑制了C3转化酶形成。图11A曲线图说明C3转化酶形成测定法的Fab2抗体#11的结果,在其它所测定的Fab2抗体中具有代表性,其结果见表6。因为甚至当各MASP-2分子被Fab2结合时,“阻断性”Fab2仅可能极微弱地抑制MASP-2功能,这在理论是可能的,因此要慎重考虑。
尽管甘露聚糖是凝集素途径的已知激活剂,但是在大鼠血清存在的抗甘露聚糖抗体还可能激活经典途径,并且通过经典途径C3转化酶产生C3b,这在理论上是可能的。然而,在本实施例所列的17个阻断性抗MASP-2Fab2中的每一个都有效地抑制了C3b产生(>95%),因此证明了该项测定法对于凝集素途径C3转化酶的特异性。
为了计算各个抗体的表观Kd,对阻断性Fab2的所有17个都进行结合测定。6个阻断性Fab2的抗天然大鼠MASP-2的抗大鼠MASP-2Fab2的结合测定的结果也见表6。图11B通过图示说明了用Fab2抗体#11的结合测定法的结果。对于其它Fab2也进行了类似的结合测定,其结果见表6。一般而言,对于6个Fab2的每一个结合“天然”MASP-2所获得的表观Kd与在C3转化酶功能测定中Fab2的IC50的对应性颇为适当。有证据表明,在激活其蛋白酶活性时,MASP-2经历了从“无活性”到“活性”形式的构象变化(Feinberg等,EMBO J 22:2348-59(2003);Gal等,J.Biol.Chem.280:33435-44(2005))。在用于C3转化酶形成测定法的正常大鼠血浆中,MASP-2主要以“无活性”酶原构象存在。相比之下,在结合测定法中,MASP-2作为与MBL一起结合到固定化甘露聚糖的复合物的组成部分存在;因此,MASP-2可能为“活性”构象(Petersen等,J.Immunol Methods 257:107-16,2001)。因此,对于这两个功能测定法中所测定的17个阻断性Fab2的每一个,可能不必预期IC50与Kd之间确切的对应性,因为在各个测定法中,Fab2可能结合不同构象形式的MASP-2。尽管如此,除了Fab2#88以外,两种测定法中所测的其它16个Fab2的每一个,IC50与表观Kd之间似乎有相当密切的对应性(参见表6)。
对用于抑制MASP-2介导的C4裂解的若干个阻断性Fab2进行了评价。图11C通过图示说明了C4裂解测定法的结果,表明用Fab2#41抑制的IC50=0.81nM(参见表6)。如图12所示,发现所有测试的Fab2都抑制C4裂解,IC50类似于C3转化酶测定法中所获得的IC50(参见表6)。
尽管甘露聚糖是凝集素途径的已知激活剂,但是大鼠血清中抗甘露聚糖抗体的存在也可能激活经典途径,从而通过C1s介导的C4裂解而产生C4b,这在理论上是可能的。然而,已经鉴定出有效抑制C4b产生(>95%)的若干种抗MASP-2Fab2,因此证明了该测定法对于MASP-2介导的C4裂解的特异性。C4,如同C3一样,含有独特的高反应性硫酯基团作为其结构部分。在该测定法中当通过MASP-2裂解C4时,C4b上的硫酯基团可与通过酯键或酰胺键固定在塑料孔底部的大分子上羟基或氨基形成共价键,因此有利于在ELISA测定法中检测出C4b。
这些研究清楚表明,对于大鼠MASP-2蛋白的高亲和性FAB2的产生,功能性地阻断C4和C3转化酶活性,从而防止了凝集素途径活化。
实施例25
本实施例描述了对若干按照实施例24所述方法产生的阻断性抗大鼠MASP-2Fab2抗体进行的表位作图。
方法:
如图13所示,使用pED4载体,在CHO细胞中表达下列都具有N端6个His标记的蛋白质:
大鼠MASP-2A,一种全长MASP-2蛋白,通过活性中心上的丝氨酸改变为丙氨酸(S613A)而失活;
大鼠MASP-2K,经改变降低了自身活化的全长MASP-2蛋白(R424K);
CUBI-II,一种仅含有CUBI、EGF样和CUBII结构域的大鼠MASP-2的N端片段;和
CUBI/EGF样,一种仅含有CUBI和EGF样结构域的大鼠MASP-2的N端片段。
按照前述方法将这些蛋白质通过镍亲和层析从培养上清液中纯化出来(Chen等,J.Biol.Chem.276:25894-02(2001))。
采用pTrxFus(Invitrogen),使含有CCPII和大鼠MASP-2丝氨酸蛋白酶结构域的C端多肽(CCPII-SP),在大肠杆菌中表达成为硫氧还蛋白融合蛋白。用Thiobond亲和树脂将蛋白质从细胞裂解物中纯化出来。硫氧还蛋白融合物配偶体由pTrxFus空载体表达作为阴性对照。
将所有重组蛋白透析到TBS缓冲液中,通过测量OD(280nm),测得其浓度。
斑点印迹法分析:
将连续稀释的上述和图13中所示5个重组MASP-2多肽(以及硫氧还蛋白多肽作为CCPII-丝氨酸蛋白酶多肽的阴性对照)点在硝酸纤维素膜上。蛋白质的点样量在5重步骤中的范围为100ng-6.4pg。在稍后的实验中,蛋白质的点样量再次在5重步骤中的范围由50ng降至16pg。该膜用5%脱脂奶粉的TBS溶液(封闭缓冲液)封闭,然后与1.0μg/ml抗MASP-2Fab2的封闭缓冲液(含有5.0mM Ca2+)一起温育。用HRP缀合的抗人Fab(AbD/Serotec;1/10,000稀释)和ECL检测试剂盒(Amersham)检测结合的Fab2。一块膜与作为阳性对照的多克隆兔抗人MASP-2Ab(参见Stover等,J Immunol 163:6848-59(1999))一起温育。在这种情况下,用HRP缀合的山羊抗兔IgG(Dako;1/2,000稀释),检测出结合的Ab。
MASP-2结合测定法
ELISA板用1.0μg/孔的重组MASP-2A或CUBI-II多肽的碳酸盐缓冲液(pH 9.0)在4℃下包被过夜。孔用1%BSA的TBS溶液封闭,然后加入连续稀释的抗MASP-2Fab2的TBS溶液(含有5.0mM Ca2+)。将所述板在室温下温育1小时。用TBS/吐温/Ca2+洗涤3次后,加入按1/10,000稀释于TBS/Ca2+的HRP缀合的抗人Fab(AbD/Serotec),将所述板再次在室温下温育1小时。用TMB过氧化物酶底物试剂盒(Biorad)检测出结合的抗体。
结果:
斑点印迹法分析的结果表明了Fab2与下表7中提供的各种MASP-2多肽的反应性。表7提供的数值表明,所需要的蛋白质点样量提供大约最大信号强度的一半。如表所示,所有多肽(仅硫氧还蛋白融合物配偶体例外)均被阳性对照Ab(多克隆抗人MASP-2血清,在兔中产生)识别。
表7:斑点印迹法中与各重组大鼠MASP-2多肽的反应性
NR=无反应。阳性对照抗体是在兔中产生的多克隆抗人MASP-2血清。
所有Fab2均与MASP-2A以及MASP-2K反应(数据未显示)。大多数Fab2识别CCPII-SP多肽但不识别N端片段。Fab2#60和Fab2#57是两个例外。Fab2#60识别MASP-2A和CUBI-II片段,但不识别CUBI/EGF样多肽或CCPII-SP多肽,这提示它能结合CUBII的表位或者跨越CUBII和EGF样结构域。Fab2#57识别MASP-2A但不识别任何所测试的MASP-2片段,这可能表明这种Fab2识别CCP1的表位。Fab2#40和#49仅结合完整的MASP-2A。在图14所示的ELISA结合测定法中,Fab2#60还结合CUBI-II多肽,虽然只具有略微较低的表观亲和性。
这些观察结果表明对于MASP-2蛋白多个区域上的独特阻断性Fab2的鉴定。
实施例26
本实施例描述了鼠肾缺血/再灌注模型中MASP-2-/-小鼠的分析法。
背景/基本原理:体温下,肾缺血-再灌注(I/R)损伤与多种临床病症有关,包括低血容量性休克、肾动脉闭塞和交叉钳夹术。
肾缺血-再灌注(I/R)是急性肾衰竭的重要原因,相关死亡率高达50%(Levy等,JAMA 275:1489-94,1996;Thadhani等,N.Engl.J.Med.334:1448-60,1996)。移植后肾衰竭是肾移植常见且危险的并发症(Nicholson等,Kidney Int.58:2585-91,2000)。目前还没有肾I/R损伤的有效疗法,血液透析是现有唯一的治疗法。肾I/R损伤的病理生理学十分复杂。最新研究表明,凝集素途径的补体活化可能对肾I/R损伤的发病机制具有重要作用(deVries等,Am.J.Path.165:1677-88,2004)。
方法:
按照实施例1所述方法产生MASP-2(-/-)小鼠,并与C57Bl/6回交至少10代。将咪唑安定(Hypnovel)(6.64mg/kg;Roche products Ltd.Welwyn Garden City,UK)注射到称重介于22-25g之间的6只雄性MASP-2(-/-)和6只野生型(+/+)小鼠腹膜内,随后通过吸入异氟烷(Abbott Laboratories Ltd.,Kent,UK)使之麻醉。之所以选择异氟烷,是因为它是温和的吸入麻醉剂,肝毒性最小;浓度调节准确,即使在长时间麻醉后,动物也能迅速恢复。之所以给予咪唑安定,是因为它在动物中产生安定麻醉的条件,这意味着只需要给予较少的异氟烷。将保暖垫(warm pad)放置在动物身体下面以便保持恒定体温。接着,沿腹中线切开,用一副牵开器使腹腔保持打开状态。彻底清除掉左右肾的肾静脉和动脉的结缔组织,使用微动脉瘤夹钳紧紧夹住肾蒂55分钟。这种一段时间的缺血最初是根据在实验室中进行的先期研究来进行的(Zhou等,J.Clin.Invest.105:1363-71(2000))。另外,在缺血滴定后选择55分钟的标准缺血时间,并且发现55分钟造成仍可逆转的稳定损伤,死亡率低,为5%以下。闭塞后,将0.4ml热的盐水(37℃)注入腹腔内,然后闭合腹部达缺血时间。取出微动脉瘤夹钳后,观察肾脏直到变色,这是血液回流到肾脏的表示。另将0.4ml热的盐水注入腹腔内,将切口缝合,之后把动物放回其笼中。取出夹钳后24小时采集尾部血样,48小时时处死小鼠,再次收集血样。
肾损伤的评价:在6只雄性MASP-2(-/-)和6只野生型(WT)(+/+)小鼠再灌注后24小时和48小时,对肾功能进行了评价。通过质谱法测定血液肌酸酐的量,这提供了可再现的肾功能指标(灵敏度<1.0μmol/L)。图15通过图示说明了在再灌注后24小时和48小时,野生型C57Bl/6对照和MASP-2(-/-)的血尿素氮清除率。如图15所示,与野生型对照小鼠相比,MASP-2(-/-)小鼠在24小时和48小时时呈现出血尿素的量显著降低,这表明在缺血再灌注损伤模型中对肾损伤的保护性功能作用。
总的来说,在手术和缺血损伤后24小时和48小时,在WT(+/+)和MASP-2(-/-)小鼠中均观察到血尿素增加。单独测定了非缺血WT(+/+)手术动物中的血尿素水平为5.8mmol/L。除了图15所提供的数据外,1只MASP-2(-/-)动物显示受到几乎完全的保护而未发生缺血损伤,24小时和48小时的值分别为6.8mmol/L和9.6mmol/L。这只动物被排除在组别分析之外作为可能的无关项,其中可能存在无缺血损伤。因此,图15所示最终分析包括5只MASP-2(-/-)小鼠和6只WT(+/+)小鼠,在MASP-2(-/-)小鼠中观察到在24小时和48小时时,血尿素在统计学上有显著降低(Student t-检验p<0.05)。这些观察结果表明抑制MASP-2活性可能将对因缺血损伤所造成的肾损伤产生保护或治疗作用。
实施例27
本实施例描述了对小鼠心肌缺血/再灌注模型中MASP-2(-/-)小鼠的分析。
背景/基本原理:
甘露糖结合凝集素(MBL)是在响应各种各样的糖结构时以免疫复合物依赖性的方式启动补体活化的循环分子。这些结构可以是感染因子(infectious agent)的成分或发生变化的内源糖部分,特别是坏死细胞、肿胀细胞或凋亡细胞内的糖部分。这些细胞死亡的形式发生在再灌注的心肌层内,其中补体活化很可能使损伤扩大到由再灌注终止缺血的那一时刻存在的损伤界限以外。尽管引人注目的证据表明,补体活化使心肌再灌注恶化,但是仍不清楚这种活化的机制,而且抑制所有的已知途径很可能具有不堪耐受的不利作用。最新研究表明活化可能包括MBL,而不是经典途径或替代放大环路(如本文中定义),因为是在MBL(A/C)-失效小鼠而不是在C1q-失效小鼠中有梗塞减少(Walsh M.C.等,Jour of Immunol.175:541-546(2005))。然而,这虽然令人鼓舞,但是这些小鼠仍然包括循环成分(例如纤维胶凝蛋白A),能够通过凝集素途径激活补体。
该研究对MASP-2(-/-)小鼠与野生型(+/+)对照进行了研究以确定MASP-2(-/-)是否对心肌缺血和再灌注损伤不大敏感。使MASP-2(-/-)小鼠遭受局部缺血,将梗塞大小与其野生型同窝小鼠的进行比较。
方法:以下方案以前述诱导缺血/再灌注损伤的方法(Marber等,J.Clin Invest.95:1446-1456(1995))为基础。
按照实施例1所述方法产生MASP-2(-/-)小鼠,并与C57Bl/6回交至少10代。7只MASP-2(-/-)小鼠和7只野生型(+/+)小鼠用氯胺酮/美托咪定(分别为100mg/kg和0.2mg/kg)麻醉,并使之仰卧在温控加热垫上以维持直肠温度在37±0.3℃。在直视下给小鼠插管,在110次/分钟的呼吸频率和225μl/分钟的潮气量下使室内空气流通(通风器-Hugo Sachs Elektronic MiniVent Type 845,Germany)。
备皮,从左腋窝到剑突前外侧皮肤切开。剖开胸大肌,从其胸骨边缘切起,并切到腋窝。胸小肌从其颅骨边缘切起,并切到尾侧。该肌肉稍后在冠状动脉闭塞期间用作覆盖心脏的肌瓣。从左肺稍靠中间到边缘的点用镊子刺入第5肋间隙和胸膜壁层的肌肉,从而避免了损伤肺或心脏。在刺穿胸膜后,小心地将镊子指向胸膜以外直指胸骨而不触及心脏,用电池驱动的烙器(Harvard Apparatus,UK)剖开胸膜和肋间肌。操作上要特别留意避免任何出血。用同一技术,将胸廓切开术延伸到腋窝中线。从其胸骨边缘切断第4肋骨后,使肋间隙扩宽直到整个心脏从基底部到顶部暴露出来。用两个小的动脉钳打开心包,围心托架(pericardial cradle)设置成使心脏稍微前移。使冠状动脉左前降支(LAD)暴露出来,然后将穿有8-0单丝缝合线的圆针在LAD下穿过。LAD的结扎部位正好位于左心房尖的尾部,沿房室嵴(atrioventricular crest)到左心室顶端线路的大约1/4。
所有实验都以不知情的方式进行,研究人员不知道各个动物的基因型。在仪器使用和外科手术完成后,允许小鼠有15分钟的平衡期。然后小鼠进行30分钟的冠状动脉闭塞与120分钟的再灌注时间。
冠状动脉闭塞和再灌注模型
用前述的悬重系统(hanging weight system)形成冠状动脉闭塞(Eckle等,Am J Physiol Heart Circ Physiol 291:H2533-H2540,2006)。将单丝结扎线的两端穿过一段长2mm的聚乙烯PE-10管中,并且用氰基丙烯酸酯胶粘到长度5-0的缝合线上。然后将缝合线牵引在2个水平安置的可移动金属棒上,缝合线的两端各附加上1g的小块。通过抬升小棒,使小块悬起,置于控制牵引力下的缝合线随着规定且恒定的压力使LAD闭塞。LAD闭塞通过处于风险的区域变苍白而得到验证,LAD灌注区的颜色由鲜红变成紫色,这就表明血流停止。通过放低小棒直到小块放到手术垫上,并且解除了结扎线的牵引力而获得再灌注。通过与用于验证闭塞相同的3个标准来验证再灌注。如果分别在冠状动脉闭塞开始时或者在再灌注15分钟内未达到所有3个标准,则小鼠不再用于进一步分析。在冠状动脉闭塞期间,通过用胸大肌瓣覆盖心脏,并且用0.9%盐水浸湿的纱布封住胸廓切开伤口,来维持心脏表面的温度和湿度。
心肌梗塞大小的测量:
通过测面积法(planometry)来测定梗塞大小(INF)和风险区域(AAR)。在静脉内注射500I.U.肝素后,使LAD再闭塞,将300μl 5%(重量/体积)伊文思蓝(Evans Blue)(Sigma-Aldrich,Poole,UK)慢慢注射到颈静脉内以显现风险区域(AAR)的轮廓。这使得染料进入左心室的非缺血区域,而缺血AAR则不染色。小鼠断颈处安乐死,快速取出心脏。将心脏在冰上冷却,并在5%琼脂糖中固定,然后切成8片厚度为800μm的横切片。将所有切片与溶于0.1M Na2HPO4/NaH2PO4缓冲液(调节至pH 7.4)的3%2,3,5-三苯基氯化四唑(Sigma Aldrich,Poole,UK)一起在37℃下温育20分钟。将切片在10%甲醛中固定过夜。将切片放在两块盖玻片之间,采用高分辨率光学扫描仪对各切片两侧进行数字成像。然后运用SigmaScan软件(SPSS,US)对数字图像进行分析。通过鉴定其颜色外观和颜色边界,在每个切片上描出梗塞区域(苍白色)的大小、左心室(LV)风险区域(红色)和正常灌注的LV带(蓝色)的轮廓。研究人员对每个切片两侧的面积进行定量测定并平均。计算每只动物的梗塞大小,用%风险区域表示。
结果:通过鉴定其颜色外观和颜色边界,在每个切片上描出梗塞区域(苍白色)的大小、LV风险区域(红色)和正常灌注的LV带(蓝色)的轮廓。研究人员对每个切片两侧的面积进行定量测定并平均。计算每只动物的梗塞大小,用%风险区域表示。图16A表示对7只WT(+/+)小鼠和7只MASP-2(-/-)小鼠的评价,以在经历上述冠状动脉闭塞和再灌注技术后,测出它们的梗塞大小。如图16A所示,MASP-2(-/-)小鼠比起野生型(+/+)小鼠,梗塞大小在统计上显著减少(p<0.05),这就表明在缺血再灌注损伤模型中具有保护心肌免受损伤的作用。图16B显示所测试的个体动物的分布,表明了对MASP-2(-/-)小鼠明显的保护作用。
实施例28
本实施例描述了MASP-2-/-在鼠黄斑变性模型中的结果。
背景/基本原理:年龄相关性黄斑变性(AMD)是工业化世界55岁以后致盲的主要原因。AMD主要以两种形式出现:新血管性(湿性)AMD和萎缩性(干性)AMD。新血管(湿性)形式占AMD相关的严重视力丧失的90%,即使仅有~20%患有AMD的个体发展成湿性形式。AMD的临床特点包括多发性脉络膜小疣、地区性萎缩(geographic atrophy)和脉络膜新血管形成(CNV)。2004年12月,美国食品和药品管理局(FDA)批准了Macugen(倍加他尼(pegaptanib)),这是一类特异性靶向并阻断血管内皮生长因子(VEGF)的作用新的眼科药物,用于治疗湿性(新血管)形式的AMD(Ng等,Nat Rev.Drug Discov 5:123-32(2006))。尽管Macugen对于小部分AMD患者代表着有前景的新的治疗选择,但是对于开发这种复杂疾病的其它治疗方法的迫切需要依然存在。多项独立路线的研究表明了补体活化在AMD发病机制中的重要作用。脉络膜新血管形成(CNV)的发病机制,是一种最严重的AMD形式,可能包括补体途径的活化。
25年前,Ryan描述了动物CNV的激光诱导的损伤模型(Ryan,S.J.,Tr.Am.Opth.Soc.LXXVII:707-145,1979)。该模型最初是用猕猴开发的,然而,此后已经应用同一技术在各种研究动物中开发出了类似的CNV模型,包括小鼠(Tobe等,Am.J.Pathol153:1641-46,1998)。在这一模型中,使用激光凝固来破坏脉胳膜基底层(Bruch's membrane),这种做法将导致形成CNV样膜。激光诱导的模型获取了人的该种疾病的重要特征(有关最新综述可参见Ambati等,Survey Ophthalmology 48:257-293,2003)。现已建立了良好的激光诱导的小鼠模型,并且在大量甚至不断增加的多个研究项目中用作实验基础。一般公认的是,激光诱导的模型与人的CNV共有足够的生物相似性,使用该模型的发病机制的临床前研究和药物抑制与人的CNV有关。
方法:按照实施例1所述方法产生MASP-2-/-小鼠,并与C57Bl/6回交10代。目前的研究对当在激光诱导的CNV的病程中对MASP-2(-/-)和MASP-2(+/+)雄性小鼠评价的结果进行了比较,激光诱导的CNV是一种在激光损伤后,通过激光扫描共焦显微镜来衡量组织损伤,并且通过ELISA测定视网膜色素上皮(RPE)/脉络膜的VEGF(一种涉及CNV的有效血管生成因子)水平,焦点集中在激光诱导的CNV体积的新血管AMD的加速模型。
诱导脉络膜新血管形成(CNV):在第0天,由对药物组分配不知情的一个人,对每只动物的双眼实施激光凝固(532nm,200mW,100毫秒,75μm;Oculight GL,Iridex,Mountain View,CA)。使用裂隙灯传递系统和盖玻片作为接触透镜,按标准化方式将激光光斑照在视神经周围。激光损伤形态终点是出现空泡,这是一种被视作与脉胳膜基底层破坏有关的迹象。详细方法和终点数据评价如下。
荧光素血管造影术:在激光凝固后1周,用照相机和成像系统(TRC 50 1A照相机;ImageNet 2.01系统;Topcon,Paramus,NJ)进行荧光素血管造影术。在腹膜内注射0.1ml2.5%荧光素钠后,用与眼底照相机镜头接触的20-D镜头抓拍照片。未参与激光凝固或血管造影术的视网膜专家以不知情的方式一次性对荧光素血管造影照片进行评价。
脉络膜新血管形成(CNV)的体积:在激光损伤后一周,摘出双眼,用4%低聚甲醛在4℃下固定30分钟。通过除去前节(anterior segment)获得眼杯,在PBS中洗涤3次,随后通过甲醇系列脱水和再水化。用缓冲液(含有1%牛血清白蛋白和0.5%Triton X-100的PBS)在室温下封闭两次30分钟后,眼杯与稀释于含有0.2%BSA和0.1%Triton X-100的PBS中的0.5%FITC-同工凝集素B4(Vector laboratories,Burlingame,CA)一起在4℃下温育过夜,FITC-同工凝集素B4能结合内皮细胞表面的末端β-D-半乳糖残基,并选择性地标记鼠血管系统。用含有0.1%Triton X-100的PBS洗涤2次后,轻轻剥离视网膜感觉神经层(neurosensory retina)并切断视神经。切开4个松驰放射状切口,将剩余的RPE-脉络膜-巩膜复合体平放在antifade培养基(Immu-Mount Vectashield Mounting Medium;Vector Laboratories)中封固,盖上盖玻片。
平放样本(Flatmount)用激光扫描共焦显微镜(TCS SP;Leica,Heidelberg,Germany)观察。通过用蓝色氩波长(488nm)激发并捕获515nm和545nm之间的发射来观测血管。对于所有成像研究都使用40X油浸物镜。从RPE-脉络膜-巩膜复合体表面获取水平光学切片(lμm步进)。可以鉴定出在与病变相连的围绕脉络膜血管网上的最深焦平面,这被认定是病变的基底。在激光定向区域与这一参照平面表面上的任何血管均被断定为CNV。各切片的图像通过数字化予以保存。用显微镜软件(TCS SP;Leica)通过计算机进行的图像分析来测定与CNV相关荧光的面积。在各个水平切片上对整个荧光面积进行汇总,来用作CNV体积的指数。由对处理组分配不知情的操作人员进行成像。
因为各个激光病变发展成CNV的可能性受它所属组别的影响(小鼠、眼和激光光斑),使用具有裂区重复测量设计的线性混合模型对平均病变体积进行比较。主区(whole plot)因子是动物所属的遗传组别,而裂区(split plot)则为眼。测得统计上的显著性水平为0.05。用适于多重比较的Bonferroni调整(Bonferroni adjustment)构建事后比较的方法。
VEGF ELISA。在通过12束激光光斑损伤后的第3天,将RPE-脉络膜复合体于裂解缓冲液(20mM咪唑HCl,10mM KCl,1mM MgCl2,10mM EGTA,1%Triton X-100,10mM NaF,1mM钼酸钠和1mM EDTA与蛋白酶抑制剂)中在冰上用超声处理15分钟。用识别所有切片变体的ELISA试剂盒(R&D Systems,Minneapolis,MN),在450-570nm(Emax;Molecular Devices,Sunnyvale,CA)下测定上清液中的VEGF蛋白水平,并针对总蛋白标准化。由不参与激光凝固、成像或血管造影术的操作人员以不知情的方式进行重复测量。VEGF数值用至少3次独立实验的平均值+/-SEM表示,采用Mann-Whitney U检验进行比较。在P<0.05时拒绝零假设。
结果:
评价VEGF水平:
图17通过图示说明了在第0天由C57Bl6野生型和MASP-2(-/-)小鼠中分离出来的RPE-脉络膜复合体的VEGF蛋白水平。如图17A所示,对VEGF水平的评价表明,在MASP-2(-/-)小鼠与C57bl野生型对照小鼠中VEGF的基线水平降低。图17B通过图示说明了激光诱导的损伤后第三天所测VEGF蛋白水平。如图17B所示,在野生型(+/+)小鼠中,在激光诱导的损伤之后的第三天,VEGF水平显著提高,这与已发表的研究(Nozaki等,Proc.Natl.Acad.Sci.USA 103:2328-33(2006))一致。然而,预料不到的是,在MASP-2(-/-)小鼠中观察到VEGF的水平非常低。
评价脉络膜新血管形成(CNV):
除了在激光诱发的黄斑变性之后VEGF水平降低以外,在激光损伤之前和之后测定了CNV面积。图18通过图示说明了在C57bl野生型小鼠和MASP-2(-/-)小鼠中,在激光诱导的损伤后的第7天测得的CNV体积。如图18所示,与野生型对照小鼠相比,MASP-2(-/-)小鼠的CNV面积在激光诱导的损伤后的第7天呈大约30%的减少。
这些观察结果表明在MASP(-/-)小鼠与野生型(+/+)对照中观察到的VEGF和CNV减少,用抑制剂阻断MASP-2可能在黄斑变性的治疗中具有保护或治疗作用。
实施例29
本实施例描述了MASP-2(-/-)在鼠单克隆抗体诱发的类风湿性关节炎模型中的结果。
背景/基本原理:类风湿性关节炎(RA)最常用的动物模型是胶原诱发的关节炎(CIA)(有关最新综述可参见Linton和Morgan,Mol.Immunol.36:905-14,1999)。胶原II型(CII)是关节基质蛋白的一种主要组分,用天然CII与佐剂免疫通过对关节软骨中与CII有交叉反应性自体免疫应答而诱发自身免疫性多关节炎。同RA一样,对CIA的易感性与某些II类MHC等位基因的表达相关联。小鼠的一些品系,包括C57Bl/6品系,对经典CIA具有抗性,因为它们缺乏适当的MHC单元型,因此不会产生高的抗CII抗体效价。然而,已经发现经静脉内或腹膜内给予小鼠4种抗II型胶原的特异性单克隆抗体合剂,可在小鼠所有品系中诱发一致的关节炎。这些致关节炎单克隆抗体(arthridogenic monoclonal antibodies)是市售的(Chondrex,Inc.,Redmond,WA)。这种CIA的被动转移模型已成功用在多个近期发表的采用C57Bl/6小鼠品系的报道之中(Kagari等,J.Immunol.169:1459-66,2002;Kato等,J.Rheumatol.30:247-55,2003;Banda等,J.Immunol.177:1904-12,2006)。下面的研究对使用CIA的被动转移模型对野生型(+/+)(WT)与MASP-2(-/-)小鼠发生关节炎的敏感性进行了比较,这两种小鼠具有共同的C57Bl/6遗传背景。
方法:
动物:按照实施例1所述方法产生MASP-2(-/-)小鼠,并与C57Bl/6回交10代。在本项研究中,使用的14只雄性和雌性C57BL/6野生型小鼠在抗体注射时为7-8周龄,10只雄性和雌性MASP-2(-/-)和野生型(+/+)C57Bl/6小鼠在抗体注射时为7-8周龄。20只小鼠用单克隆抗体合剂注射,得到20只一致应答小鼠(两组各10只)。将动物(10/组)按5只动物/笼关养,在开始研究前使之适应5-7天。
在第0天和第1天,给小鼠静脉内注射单克隆抗体合剂(Chondrex,Redmond WA)(5mg)。试验药物是得自Chondrex公司的单克隆抗体+LPS。在第2天,经腹膜内给予小鼠LPS。在第0天、第2天、第4天、第6天、第8天、第10天、第12天和(结束前)第14天,给小鼠称重。在第14天,小鼠用异氟烷麻醉,最终放血用于血清。收集血液后,小鼠处安乐死,取下带膝的前后肢,放入福尔马林中留待将来处理。
处理组:
第1组(对照):4只小鼠,品系C57/BL/6WT(+/+);
第2组(试验):10只小鼠,品系C57/BL/6WT(+/+)(接受mAb混合物加上LPS);和
第3组(试验):10只小鼠,品系C57/BL/MASP-2KO/6Ai(-/-)(接受mAb混合物加上LPS)
用以下评分系统每日评价临床关节炎评分:
0=正常;1=1只后爪或前爪关节染病;2=2只后爪或前爪关节染病;3=3只后爪或前爪关节染病;4=中度(红斑和中度肿胀或4节指趾关节染病);5=严重(弥散性红斑和整只爪严重肿胀,不能伸缩指趾)。
结果:
图19显示用于在长达两周时间内绘制平均每日临床关节炎评分曲线图的分组数据。在未接受CoL2MoAb处理的对照组中未观察到临床关节炎评分。从第9天到第14天,MASP(-/-)小鼠的临床关节炎评分较低。总体临床关节炎评分与曲线下面积(AUC)分析表明MASP-2(-/-)组相对于WT(+/+)小鼠降低了21%。然而,前述C57Bl6小鼠本底无法提供完整的总体关节炎临床评分。由于发病率低,分组数少,故虽然是肯定的趋势,所提供的数据却只是倾向(p=0.1),在统计上没有显著性p<0.05的水平。可能需要处理组中额外的动物来表示统计上的显著性。由于关节炎的发病率降低,所以评价了染病爪评分用于说明严重程度。在任一MASP-2(-/-)小鼠中未观察到临床关节炎评分>3的单一发病情况,而在30%WT(+/+)小鼠中则观察到,这进一步表明(1)关节炎的严重程度可能与补体途径活化有关,(2)阻断MASP-2可能对关节炎具有有益效果。
实施例30
本实施例证实,小的甘露糖结合凝集素结合蛋白(MAp19或sMAP)是MASP-2依赖性补体活化的抑制剂。
背景/基本原理:
摘要:
甘露糖结合凝集素(MBL)和纤维胶凝蛋白是在先天免疫中起作用的模式识别蛋白,并且通过MBL相关的丝氨酸蛋白酶(MASP)触发凝集素补体途径活化。在凝集素途径活化时,MASP-2裂解C4和C2。小的MBL相关蛋白(sMAP),一种截短形式的MASP-2,同样与MBL/纤维胶凝蛋白-MASP复合物缔合。为了阐明sMAP的作用,我们通过定向破坏sMAP特异性外显子产生了sMAP缺陷型(sMAP-/-)小鼠。因为基因受到破坏,MASP-2的表达水平在sMAP-/-小鼠中也降低。在缺陷型血清中重组sMAP(rsMAP)和重组MASP-2(rMASP-2)再构成MBL-MASP-sMAP复合物时,这些重组体与MBL的结合是竞争性的,MBL-MASP-sMAP复合物的C4裂解活性通过加入rMASP-2而得到恢复,然而加入rsMAP削弱了活性。因此,MASP-2对于C4活化是必不可少的,sMAP在凝集素途径活化中起着调节作用。
绪论:
补体系统介导蛋白酶解的连锁反应和蛋白质复合物的装配,作为先天性免疫系统和适应性免疫系统两者的一部分在生物防御中起着主要作用。哺乳动物补体系统由三条活化途径组成,即经典途径、替代途径和凝集素途径(Fujita,Nat.Rev.Immunol.2:346-353(2002);Walport,N Engl J Med 344:1058-1066(2001))。凝集素途径提供针对入侵病原体的第一线防御。这个途径的病原体识别成分甘露糖结合凝集素(MBL)和纤维胶凝蛋白,能结合细菌、病毒和寄生虫表面的一系列糖,并激活MBL相关的血清蛋白酶(MASP),从而触发下游反应级联。将缺乏MBL与各种感染性疾病(特别是在适应性免疫系统建立之前的低龄儿童时期)易感性增加联系在一起的许多临床研究强调凝集素途径对于先天免疫防御的重要性(Jack等,Immunol Rev180:86-99(2001);Neth等,Infect Immun68:688-693(2000);Summerfield等,Lancet 345:886-889(1995);Super等,Lancet 2:1236-1239(1989))。然而,凝集素途径也是造成补体不良活化的原因,这种补体的不良活化在包括心脏和肾脏缺血/再灌注损伤等许多病理病症中参与炎症和组织损伤(de Vries等,Am J Pathol 165:1677-1688(2004);Fiane等,Circulation108:849-856(2003);Jordan等,Circulation 104:1413-1418(2001);Walsh等,J Immunol175:541-546(2005))。
正如上面提及的一样,凝集素途径包括通过MBL和纤维胶凝蛋白的糖识别(Fujita等,Immunol Rev 198:185-202(2004);Holmskov等,Annu Rev Immunol 21:547-578(2003);Matsushita和Fujita,Immunobiology 205:490-497(2002)),这些凝集素与以下成分形成复合物:MASP-1(Matsushita和Fujita,J Exp Med 176:1497-1502(1992);Sato等,Int Immunol6:665-669(1994);Takada等,Biochem Biophys Res Commun 196:1003-1009(1993))、MASP-2(Thiel等,Nature 386:506-510(1997),MASP-3(Dahl等,Immunity 15:127-135(2001))和MASP-2的截短蛋白(小MBL相关蛋白;sMAP或MAp19)(Stover等,J Immunol 162:3481-3490(1999);Takahashi等,Int Immunol 11:859-863(1999))。MASP家族成员由6个结构域组成,2个C1r/C1s/Uegf/骨形成蛋白(CUB)结构域,1个表皮生长因子(EGF)样结构域,2个补体调控蛋白(CCP)或短共有重复序列(short consensus repeat,SCR)结构域和1个丝氨酸蛋白酶结构域(Matsushita等,Curr Opin Immunol 10:29-35(1998))。MASP-2和sMAP可由单个结构基因通过可变剪接而产生,sMAP由第一CUB(CUB1)结构域、EGF样结构域和sMAP特异性外显子编码的C端上的额外4个氨基酸组成。MASP-1和MASP-3同样由单个基因通过可变剪接产生(Schwaeble等,Immunobiology 205:455-466(2002)。当MBL和纤维胶凝蛋白与微生物表面的糖结合时,酶原形式的MASP便在第二CCP和蛋白酶结构域之间进行切割,产生由2个多肽组成的活性形式,称为重(H)链和轻(L)链,因此获得针对补体成分的蛋白水解活性。已积累的证据表明,MASP-2切割C4和C2(Matsushita等,J.Immunol 165:2637-2642(2000)),这导致形成C3转化酶(C4bC2a)。我们假定,MASP-1直接切割C3,随后激活放大环路(Matsushita和Fujita,Immunobiology 194:443-448(1995)),但是这个功能是有争议的(Ambrus等,J.Immunol 170:1374-1382(2003))。尽管MASP-3在L链同样含有丝氨酸蛋白酶结构域,并且具有针对合成底物的酶解活性(Zundel等,J Immunol 172:4342-4350(2004)),但是还没有鉴定出它的生理底物。仍不清楚缺乏丝氨酸蛋白酶结构域的sMAP的功能。
在本研究中,为了阐明sMAP在凝集素补体途径活化中的作用,我们破坏了编码sMAP的C端4个氨基酸残基(EQSL)的sMAP特异性外显子,产生了sMAP-/-小鼠。我们在此首次报道了sMAP下调凝集素途径活化的能力。
材料与方法
小鼠
构建了含有外显子1-4和129/Sv小鼠MASP-2基因外显子6部分及新霉素抗性基因表达盒而不是外显子5的打靶载体(图20A)。将DT-A基因插入该载体的3'端,并插入3个lox p位点以便将来实施有条件打靶以定向除去新霉素表达盒和启动子区。打靶载体通过电穿孔进入129/Sv ES细胞。将靶向的ES克隆微注射到C57Bl/6J胚泡内,将胚泡植入代孕ICR母鼠的子宫内。使雄性嵌合小鼠与雌性C57BL/6J小鼠交配以产生杂合(+/-)小鼠。用图20A中所示的探针,对用BamH I消化的尾部DNA进行DNA印迹分析,筛选出杂合(+/-)小鼠。DNA印迹分析显示在得自杂合(+/-)小鼠的DNA中有6.5kbp和11kbp条带(图20B)。将杂合(+/-)小鼠与C57BL/6J小鼠回交。为了获得纯合(-/-)小鼠,使杂合(+/-)小鼠互交。通过基于PCR的尾部DNA的基因分型分析来鉴定纯合(-/-)小鼠(C57BL/6J背景)。用外显子4特异性和neo基因特异性有义引物与外显子6特异性反义引物的混合物进行PCR分析。得自纯合(-/-)小鼠的DNA产生了单一的1.8kbp条带(图20C)。在所有实验中,按照Fukushima Medical University的动物实验指导方针,来使用8-12周龄的小鼠。
RNA印迹分析
通过电泳从野生型(+/+)和纯合(-/-)小鼠肝中分离出Poly(A)+RNA(1μg),转移到尼龙膜上,与对sMAP、MASP-2H链、MASP-2L链或neo基因特异性的32P标记的cDNA探针杂交。剥离出同一膜,与对甘油醛-3-磷酸脱氢酶(GAPDH)特异性的探针再杂交。
定量RT-PCR
用LightCycler系统(Roche Diagnostics)进行实时PCR。由得自野生型(+/+)和纯合(-/-)小鼠肝的60ng poly(A)+RNA合成的cDNA用作实时PCR的模板,对MASP-2H链和L链及sMAP的cDNA片段进行扩增和监测。
免疫印迹法
在还原条件下将样品在10%或12%SDS-聚丙烯酰胺凝胶上进行电泳,将蛋白质转移到聚偏二氟乙烯(PVDF)膜上。用针对MASP-1的L链产生的抗MASP-1抗血清或用针对得自MASP-2H链的肽产生的抗MASP-2/sMAP抗血清,检测膜上的蛋白质。
检测MBL-MASP-sMAP复合物中的MASP和sMAP
将小鼠血清(20μl)加到含有0.1%(重量/体积)BSA(TBS-Ca2+/BSA)的480μl TBS-Ca2+缓冲液(20mM Tris-HCl(pH 7.4),0.15M NaCl和5mM CaCl2)中,与TBS-Ca2+/BSA缓冲液中的40μl 50%甘露聚糖-琼脂凝胶浆(Sigma-Aldrich,St.Louis,MO)在4℃下温育30分钟。温育之后,各凝胶用TBS-Ca2+缓冲液洗涤,将用于SDS-PAGE的样品缓冲液加到凝胶中。将凝胶煮沸,对上清液进行SDS-PAGE,随后用免疫印迹法检测MBL复合物中的MASP-1、MASP-2和sMAP。
C4沉积测定法
用TBS-Ca2+/BSA缓冲液将小鼠血清稀释至100μl。将稀释的样品加到甘露聚糖包被的微量滴定孔中,在室温下温育30分钟。各孔用冰冷的洗涤缓冲液(含有0.05%(体积/体积)吐温20的TBS-Ca2+缓冲液)洗涤。洗涤后,将人C4加到每孔中,在冰上温育30分钟。各孔用冰冷的洗涤缓冲液洗涤,将HRP缀合的抗人C4多克隆抗体(Biogenesis,Poole,England)加到各孔中。在37℃下温育30分钟后,各孔用洗涤缓冲液洗涤,将3,3',5,5'-四甲基联苯胺(TMB)溶液加到每孔中。显色后,加入1M H3PO4,在450nm下测量吸光度。
C3沉积测定法
将小鼠血清用含有0.1%(重量/体积)HSA的BBS缓冲液(4mM巴比妥,145mM NaCl,2mM CaCl2和1mM MgCl2,pH 7.4)稀释至100μl。将稀释的样品加到甘露聚糖包被的微量滴定孔中,在37℃下温育1小时。各孔用洗涤缓冲液洗涤。洗涤后,将HRP缀合的抗人C3c多克隆抗体(Dako,Glostrup,Denmark)加到每孔中。在室温下温育1小时后,各孔用洗涤缓冲液洗涤并将TMB溶液加到每孔中。按上述方法测定颜色。
重组体
按前述方法制备重组小鼠sMAP(rsMAP)、rMASP-2和其在丝氨酸蛋白酶结构域的活性位点丝氨酸残基被取代为丙氨酸残基的无活性小鼠MASP-2突变型(MASP-2i)(Iwaki和Fujita,2005)。
MBL-MASP-sMAP复合物的重构
将纯合(-/-)小鼠血清(20μl)和不同量的MASP-2i和/或rsMAP在总体积为40μl的TBS-Ca2+缓冲液中在冰上温育过夜。将混合物与甘露聚糖-琼脂凝胶浆一起温育,按照“检测MBL-MASP-sMAP复合物中的MASPs和sMAP”中所述方法检测结合到凝胶上MBL-MASP复合物中的MASP-2i和rsMAP。
C4沉积活性的重构
将纯合(-/-)小鼠血清(0.5μl)和不同量的rMASP-2和/或rsMAP在总体积为20μl的TBS-Ca2+中在冰上温育过夜。混合物用80μl TBS-Ca2+/BSA缓冲液稀释,并加到甘露聚糖包被的孔中。按照“C4沉积实验”中所述方法进行之后的所有步骤。
结果
图20:sMAP基因的定向破坏。(A)MASP-2/sMAP基因、打靶载体和靶向等位基因的部分限制图谱。sMAP特异性外显子(外显子5)被neo基因表达盒置换。(B)从雄性嵌合小鼠与雌性C57BL/6J小鼠交配得到子代的基因组DNA的DNA印迹分析。尾部DNA用BamH I消化,与(A)中所示探针杂交。由野生型等位基因得到11kbp条带,从靶向等位基因得到6.5kbp条带。(C)PCR基因分型分析。用外显子4特异性和neo基因特异性有义引物与外显子6特异性反义引物的混合物,对尾部DNA进行了分析。从野生型等位基因得到2.5kbp条带,从靶向等位基因得到1.8kb条带。
图21:sMAP和MASP-2mRNA在纯合(-/-)小鼠中的表达。(A)RNA印迹分析。使得自野生型(+/+)和纯合(-/-)小鼠肝的Poly(A)+RNA进行电泳,转移到尼龙膜上,与对sMAP、MASP-2H链、MASP-2L链或neo基因特异性的32P标记的探针进行杂交。在纯合(-/-)小鼠中观察到对neo特异性的条带(2.2kb)。(B)定量RT-PCR。用LightCycler仪器(Roche Diagnostics)通过实时PCR使MASP-2H链和L链及sMAP cDNA片段扩增。从得自野生型(+/+)和纯合(-/-)小鼠肝的poly(A)+RNA合成的cDNA用作模板。所示数据是两次实验的平均值。
图22:纯合(-/-)小鼠血清中缺乏MASP-2。(A)小鼠血清中的MASP-2和sMAP的免疫印迹法。野生型(+/+)或纯合(-/-)小鼠血清(2μl)进行了免疫印迹法,并用抗MASP-2/sMAP抗血清进行了检测。(B)MBL-MASP-sMAP复合物中MASP和sMAP的检测。按照材料与方法中所述,将小鼠血清与甘露聚糖-琼脂糖凝胶一起温育,检测出结合到凝胶上的MBL复合物中的sMAP、MASP-1和MASP-2。
图23:在纯合(-/-)小鼠血清中,C4和C3裂解减少。(A)C4沉积在甘露聚糖包被的孔中。将小鼠血清稀释2倍,在甘露聚糖包被的孔中在室温下温育30分钟。洗涤各孔后,将人C4加到每孔中,在冰上温育30分钟。用HRP缀合的抗人C4多克隆抗体,对沉积在孔中的人C4的量进行了测定。(B)C3沉积在甘露聚糖包被的孔中。将稀释的小鼠血清加到甘露聚糖包被的孔中,在37℃下温育1小时。用HRP缀合的抗人C3c多克隆抗体检测出在孔中的内源C3沉积。
图24:sMAP和MASP-2与MBL的竞争性结合。(A)纯合(-/-)小鼠血清中MBL-MASP-sMAP复合物的重构。将MASP-2i和/或rsMAP(4μg)与纯合(-/-)小鼠血清(20μl)一起温育。将混合物再与甘露聚糖-琼脂糖凝胶一起温育,通过免疫印迹法,在结合到凝胶上的级分中检测出rsMAP和MASP-2i。(B)将不同量的MASP-2i(0-5μg)和恒定含量的rsMAP(5μg)与纯合(-/-)小鼠血清(20μl)一起温育,再与甘露聚糖-琼脂糖凝胶一起温育。(C)恒定量的MASP-2i(0.5μg)和不同量的rsMAP(0-20μg)与纯合(-/-)小鼠血清(20μl)一起温育。(D)不同量的rsMAP(0-20μg)与野生型(+/+)小鼠血清(20μl)一起温育。
图25:通过加入rMASP-2重新恢复C4沉积活性。将不同量的rsMAP(0-5μg)(A)或rMASP-2(0-1.5μg)(B)与总体积为20μl的TBS-Ca2+缓冲液中的0.5μl纯合(-/-)小鼠血清在冰上温育过夜。然后混合物用80μl TBS-Ca2+/BSA缓冲液稀释,再加到甘露聚糖包被的孔中,测得C4沉积在孔中的量。
图26:通过加入sMAP降低C4沉积活性。(A)将rMASP-2(1μg)和不同量的rsMAP(0-0.5μg)与0.5μl纯合(-/-)小鼠血清一起温育。将混合物加到甘露聚糖包被的孔中,测得C4沉积在孔中的量。(B)将rsMAP(0-0.7μg)与野生型血清(0.5μl)一起温育,测得C4沉积在甘露聚糖包被的孔中的量。
结果:
sMAP和MASP-2在纯合(-/-)小鼠中的表达
为了阐明sMAP在体内的作用,我们建立了缺乏sMAP的基因靶向小鼠。构建了打靶载体以便用新霉素抗性基因表达盒置换对sMAP特异性的外显子(外显子5)(图20A)。将阳性ES克隆注射到C57BL/6胚泡,使建立者嵌合体(founder chimera)与C57BL/6J雌性交配。得自野灰色幼鼠(agouti-color pup)尾部DNA的DNA印迹分析显示靶向等位基因的胚遗传(图20B)。用图20A中所示的探针,通过用BamH I消化的尾部DNA进行的DNA印迹分析,筛选出杂合(+/-)小鼠。DNA印迹分析显示得自杂合(+/-)小鼠的DNA的6.5kbp和11kbp条带(图20B)。使杂合(+/-)小鼠与C57BL/6J小鼠回交。为了获得纯合(-/-)小鼠,使杂合(+/-)小鼠互交。通过对尾部DNA的基于PCR的基因分型,鉴定出产生单一1.8kbp条带的纯合(-/-)小鼠(C57BL/6J背景)(图20C)。
纯合(-/-)小鼠正常发育,在体重上与野生型(+/+)小鼠没有显著差异。两者之间在形态学上也没有差异。在RNA印迹分析中,在野生型(+/+)小鼠中,sMAP特异性的探针检测到单一的0.9kb条带,而在纯合(-/-)小鼠中没有检测出特异性条带(图21A)。使用对MASP-2H链或L链特异性的探针时,在野生型(+/+)小鼠中检测出若干条特异性条带,与之前报道的一样(Stover等,1999),H链特异性探针也检测出sMAP特异性条带。然而,在纯合(-/-)小鼠中,相应的条带非常弱,并检测到几条额外的条带。我们还进行了定量RT-PCR分析,以检查sMAP和MASP-2mRNA的表达水平。在纯合(-/-)小鼠中,sMAP mRNA的表达被完全破坏,MASP-2的表达也明显降低:通过实时PCR定量测定,大约为野生型(+/+)小鼠的H链和L链两条链表达的2%(图21B)。此外,我们还研究了在蛋白质水平上的MASP-2表达。通过免疫印迹法,在纯合(-/-)小鼠血清既未检出sMAP也未检出MASP-2(图22A)。在纯合(-/-)小鼠血清与甘露聚糖-琼脂糖凝胶一起温育后,在结合到凝胶上的级分中未检出sMAP和MASP-2,尽管在复合物中检测到MASP-1(图22B)。
在纯合(-/-)小鼠血清中通过凝集素途径的C4和C3的裂解活性
当将纯合(-/-)小鼠血清在甘露聚糖包被的孔中温育,人C4沉积在孔中的量大约是从1/400至1/50稀释范围的正常血清的20%(图23A)。我们还研究了纯合(-/-)小鼠血清中凝集素途径的C3沉积活性。将小鼠血清加到甘露聚糖包被的孔中,测得内源C3沉积在孔中的量。该含量在缺陷型血清中减少,是1/10稀释的正常血清的21%(图23B)。
纯合(-/-)小鼠血清中MBL-MASP-sMAP复合物的重构
当将重组小鼠sMAP(rsMAP)或无活性小鼠MASP-2突变型(MASP-2i)加到纯合(-/-)小鼠血清中,两个重组体都能够结合MBL(图24A,泳道3和4)。当rsMAP和MASP-2i与血清同时温育时(图24A,泳道5),在MBL-MASP-sMAP复合物检测到两个重组体。然而,结合到复合物上的sMAP的量小于当仅rsMAP与血清温育的量。于是我们进一步研究了sMAP和MASP-2与MBL的竞争性结合。将恒定量的rsMAP和不同量的MASP-2i加到缺陷型血清中。rsMAP的结合以剂量依赖性方式随着MASP-2i量的增加而减少(图24B)。相反,结合到MBL上的MASP-2i的量随着rsMAP的加入而减少(图24C)。当将rsMAP加到野生型血清中时,结合到MBL的内源sMAP和MASP-2两者都以剂量依赖性方式减少(图24D)。
纯合(-/-)小鼠血清中C4沉积活性的重构
我们用重组体进行了在甘露聚糖包被孔中C4沉积的重构实验。当将rsMAP加到缺陷型血清中时,C4沉积的量实际上以剂量依赖性方式减少至基础水平(图25A)。当将rMASP-2加到血清中时,C4的量以剂量依赖性方式最多恢复至野生型血清的46%,并达到平台期(图25B)。接着,我们研究了sMAP对C4沉积的作用。当将恒定量的rMASP-2和不同量的rsMAP加到缺陷型血清中时,C4的沉积量以剂量依赖性方式随着rsMAP的加入而减少(图26A),将rsMAP加入到野生型血清中同样也减少C4沉积量(图26B),这就表明sMAP在凝集素途径活化中起着调节作用。
讨论
我们通过定向破坏sMAP特异性外显子从而产生了sMAP-/-小鼠。MASP-2的表达水平在这些小鼠的mRNA和蛋白质水平上也极大地降低(图21和图22)。用MASP-2探针的RNA印迹分析显示得自sMAP-/-小鼠的poly(A)+RNA中仅有额外条带,这表明了MASP-2基因的正常剪接被sMAP基因的打靶所改变,因此,MASP-2的表达水平明显降低。结果,与正常血清中的相比,在缺陷型血清中,经MBL-MASP复合物裂解的C4减少大约80%(图23A)。重构实验中,C4裂解活性通过加入rMASP-2而不是rsMAP而得到恢复(图25)。缺陷型血清中所观察到的C4沉积的减少可能是由MBL-MASP复合物中缺乏MASP-2所引起的(图22B)。因此,显而易见的是MASP-2对C4通过MBL-MASP复合物的活化是必需的。然而,rMASP-2的加入并不完全恢复裂解活性,而且C4的沉积达到了平台期。与之前的报道一样(Cseh等,J Immunol 169:5735-5743(2002);Iwaki和Fujita,J Endotoxin Res11:47-50(2005)),大多数rMASP-2在纯化过程中通过自身活化而转变成活性形式,一些则丧失了蛋白酶活性。因为MASP-2的活性或无活性状态对其与MBL的缔合没有重要影响(Zundel等,J Immunol 172:4342-4350(2004)),很可能丧失其蛋白酶活性的rMASP-2结合至MBL上,竞争性妨碍了活性形式的结合,从而导致了C4沉积的不完全恢复。凝集素途径的C3裂解活性在缺陷型血清中也同样减弱(图23B)。C3沉积量的减少大概是由C3转化酶活性水平非常低所致,C3转化酶由通过MASP-2产生的C4b和C2a片段组成。
MASP和sMAP各自缔结成同型二聚体,所形成的复合物通过其N端CUB和EGF样结构域与MBL或L-纤维胶凝蛋白结合(Chen和Wallis,J Biol Chem 276:25894-25902(2001);Cseh等,J Immunol 169:5735-5743(2002);Thielens等,J Immunol 166:5068-5077(2001);Zundel等,J Immunol 172:4342-4350(2004))。sMAP和MASP-2的CUB1-EGF-CUB2节段的晶体结构揭示了其同型二聚体结构(Feinberg等,EMBO J 22:2348-2359(2003);Gregory等,J Biol Chem 278:32157-32164(2003))。MBL的胶原样结构域参与了与MASP的缔合(Wallis和Cheng,J Immunol 163:4953-4959(1999);Wallis和Drickamer,J Biol Chem 279:14065-14073(1999)),引入该结构域的一些突变使得MBL与MASP-1和MASP-2的CUB1-EGF-CUB2节段的结合减弱(Wallis和Dodd,J Biol Chem 275:30962-30969(2000))。MASP-2的结合部位与MASP-1/3的结合部位重叠但不相同(Wallis等,J Biol Chem279:14065-13073(2004))。尽管还没有鉴定出MBL的sMAP结合部位,但是sMAP和MASP-2的结合部位大概是相同的,因为CUB1-EGF区与sMAP和MASP-2中的相同。因此,有理由认为sMAP和MASP-2在MBL-MASP-sMAP复合物的重构中相互竞争以结合MBL(图24)。sMAP对于MBL的亲和性比MASP-2的低(Cseh等,J Immunol 169:5735-5743(2002);Thielens等,J Immunol 166:5068-5077(2001))。仍然没有测得小鼠血清中sMAP的浓度。然而,如图22A所示,野生型血清中sMAP的量比MASP-2中的大得多。因此,sMAP能够占据MASP-2/sMAP结合部位,并妨碍MASP-2与MBL结合,因此,MBL-MASP复合物的C4裂解活性降低。sMAP在凝集素途径中的调节机制有待研究。尚不清楚sMAP是在补体活化之前还是在之后起调节作用。sMAP可能防止在微生物感染之前MBL-MASP复合物随意活化,或者抑制凝集素途径一经激活后发生过度活化。在凝集素途径中存在另外的潜在调节因子。MASP-3也是MASP-2结合MBL的竞争者,并下调MASP-2的C4裂解活性和C2裂解活性(Dahl等,Immunity 15:127-135(2001))。尽管尚未研究sMAP与MASP-3之间的相互作用,但是很可能它们能够协同下调凝集素途径的活化。
在本报告中我们证实了sMAP和MASP-2竞争结合到MBL上,且sMAP具有下调凝集素途径的能力,该途径被MBL-MASP复合物激活。有理由认为,sMAP还调节由纤维胶凝蛋白-MASP复合物激活的凝集素途径的另一条路线。MASP-2和sMAP还竞争结合小鼠纤维胶凝蛋白A,并下调纤维胶凝蛋白A-MASP复合物的C4裂解活性(Y Endo等,准备中)。最近报道了对MBL失效小鼠的研究(Shi等,J Exp Med 199:1379-1390(2004))。MBL失效小鼠在MBL凝集素途径中不具备C4裂解活性,易受金黄色葡萄球菌感染。在本研究中,同样缺失MASP-2的sMAP-/-小鼠显示,除在凝集素途径中的C4裂解活性以外,C3裂解活性还有降低。因为sMAP缺陷型小鼠的调理活性受损,所以它们易受细菌感染。对sMAP缺陷型小鼠的进一步研究将阐明凝集素途径在针对感染性疾病的保护中的功能。
另一个重要的发现是,将rsMAP加到正常血清中导致C4活化降低(图26B)。已经证实凝集素途径在若干器官中调节炎症和组织损伤(de Vries等,Am J Pathol 165:1677-1688(2004);Fiane等,Circulation 108:849-856(2003);Jordan等,Circulation 104:1413-1418(2001);Walsh等,J Immunol 175:541-546(2005))。在进行胸腹主动脉动脉瘤治疗的MBL缺陷型患者中,补体没有被激活,促炎标记的水平在手术后降低(Fiane等,Circulation108:849-856(2003))。已积累的证据证实了在各种血管床的缺血和再灌注等病症期间MBL可能的病理生理作用。因此,特异性阻断MBL或抑制凝集素补体途径可能代表着用于防止缺血/再灌注有关损伤的治疗相关性策略。因此,sMAP可能是这类抑制剂中的一个侯选者,因为它用作凝集素途径活化的弱化子(attenuator)。
实施例31
本实施例证明MASP-2负责C3的C4旁路活化。
背景/基本原理:最近,有研究表明抑制替代途径保护肾免于缺血性急性衰竭(Thurman等,J.Immunol 170:1517-1523(2003))。本文所示数据意味着凝集素途径指导替代途径活化,替代途径进而又协同放大补体活化。我们假设,凝集素途径的瞬时抑制还可影响替代途径活化,因此在限制补体介导的移植物损伤和炎症时,可改善器官移植的长期结果,并且可缓和不必要的针对移植物的适应性免疫应答的诱导,降低通过适应性免疫系统所致的继发性移植物排斥的风险。这得到了最新临床数据的支持,这些数据表明由于遗传的MBL缺陷(存在于大约30%的人群中)所导致的部分受损的凝集素途径,与人的肾同种异体移植物存活率提高有关(Berge,Am J Transplant5:1361-1366(2005))。
采用基因靶向小鼠品系,在一过性肠缺血和肌肉缺血模型中,充分证明了补体成分C3和C4参与缺血-再灌注(I/R)损伤(Weiser等,J Exp Med 183:2342-2348(1996);Williams等J Appl Physiol86:938-42,(1999))。还充分证明了C3在肾I/R损伤和继发性移植物排斥中具有显著作用(Zhou等,J Clin Invest105:1363-1371(2000);Pratt等,Nat Med 8:582-587(2002);Farrar等,Am J.Pathol 164:133-141(2004))。然而,预料不到的是在已公布的小鼠肾同种异体移植物排斥模型中没有观察到C4缺陷型的表型(Lin,2005,待发表)。然而,随后对这些C4缺陷型小鼠的血清和血浆的分析表明,这些小鼠保持残留的功能活性,表现在C3的LP依赖性裂解和进一步的下游补体活化(参见图27C)。
功能性C4旁路(和C2旁路)的存在是一些研究人员之前描述过的现象(但没有充分表征)(Miller等,Proc Natl Acad Sci 72:418-22(1975);Knutzen Steuer等,J Immunol143(7):2256-61(1989);Wagner等,J Immunol 163:3549-3558(1999),与在C4(和C2)缺陷型血清中的不依赖替代途径的C3更新有关。
方法:凝集素途径和经典途径对C3沉积的作用。将小鼠血浆(用EGTA/Mg2+作为抗凝血剂)在4.0mM巴比妥、145mM NaCl、2.0mM CaCl2、1.0mM MgCl2(pH 7.4)中稀释并再钙化,然后加到甘露聚糖(如图27A和27C所示)或酵母聚糖(如图27B所示)包被的微量滴定板,在37℃下温育90分钟。板用10mM Tris-Cl、140mM NaCl、5.0mM CaCl2、0.05%吐温20(pH 7.4)洗涤3次,然后用抗小鼠C3c抗体测得C3b沉积。
结果:图27A-C中所示结果代表着3次独立实验。当在用免疫球蛋白复合物而不是甘露聚糖或酵母聚糖包被的孔中使用同一血清时,在WT(+/+)小鼠血清和合并的MASP-2(-/-)血清中观察到C3b沉积和B因子裂解,但在C1q耗尽血清未观察到(数据未显示)。这就表明了当通过CP活性提供最初的C3b时,在MASP-2-/-血清中可以恢复替代途径活化。图27C表示预料不到的观察结果,即在C4(-/-)缺陷型血浆中可以凝集素途径依赖的方式有效地激活C3。这种“C4旁路”通过血浆与可溶性甘露聚糖或甘露糖预温育抑制凝集素途径活化而被完全破坏。
可以观察到,C3b沉积在甘露聚糖和酵母聚糖上严重危及MASP-2(-/-)缺陷型小鼠的安全,即使在按照以前已发表论文的实验条件下,所述论文认为替代途径活化应为所有三条途径所容许。如图27A-C中所示,MASP-2(-/-)缺陷型小鼠血浆不通过凝集素途径激活C4,不裂解C3,既不通过凝集素途径也不通过替代途径。因此我们假设MASP-2是这条C4旁路所必需的。Teizo Fujita教授最近报道了在鉴定很可能参与凝集素途径依赖性C4旁路的成分的进一步进展。与Fujita's MASP-1/3缺陷型小鼠品系杂交的C4缺陷型小鼠的血浆丧失了C4缺陷型血浆通过凝集素途径裂解C3的残留能力。这通过将重组MASP-1加到C4和MASP-1/3缺陷型的组合血浆中而得以恢复(Takahashi,Mol Immunol 43:153(2006),这表明MASP-1参与了在C4不存在下裂解C3的凝集素途径衍生的复合物的形成(重组MASP-1不裂解C3,但它却裂解C2;Rossi等,J Biol Chem 276:40880-7(2001);Chen等,J Biol Chem279:26058-65(2004))。我们观察到MASP-2是形成该旁路所必需的。
尽管需要更多的功能和定量参数以及组织学研究来巩固这一初步研究,但是其初步结果为通过凝集素途径的补体活化是造成肾I/R损伤的病理生理学的重要原因的假设提供了强有力的支持,正如MASP-2-/-小鼠显示的肾功能更快恢复一样。
实施例32
本实施例证明,凝血酶激活能够在生理条件下在凝集素途径激活之后发生,并且证明MASP-2牵涉的程度。在正常大鼠血清中,凝集素途径的激活导致与补体活化(以C4沉积来评定)同时发生的凝血酶激活(以凝血酶沉积来评定)。如在图28A和图28B中可见,这个系统中的凝血酶激活由MASP-2封闭性抗体(Fab2形式)抑制,显示出类似补体活化的曲线(图28A)的抑制浓度-应答曲线(图28B)。这些数据暗示,在外伤中发生时的凝集素途径的激活将在完全依赖于MASP-2的过程中导致补体系统和凝血系统两者的激活。根据推理,MASP-2封闭性抗体在过度系统性凝血例如弥散性血管内凝血的减轻病例中可证明有效,过度系统性凝血是在主要外伤病例中导致死亡率的标志之一。
实施例33
本实施例提供在MASP-2-/-缺陷小鼠和MASP-2+/+充足小鼠中使用弥散性血管内凝血("DIC")的局部Schwartzman反应模型以评估凝集素途径在DIC中的作用所产生的结果。
背景/基本原理:
如上文所述,MASP-2的阻断抑制凝集素途径激活并且减少过敏毒素C3a和C5a两者的生成。在体外,C3a过敏毒素能够显示为强有力的血小板凝聚体,但它们的体内牵涉较少被明确定义,并且伤口修复中的血小板物质和纤溶酶的释放可仅次要地牵涉补体C3。在本实施例中,在MASP-2(-/-)和WT(+/+)小鼠中分析了凝集素途径的作用,以便解决C3激活的延时上升是否为引起弥散性血管内凝血所必需。
方法:
如实施例27中所述产生用于本研究的MASP-2(-/-)小鼠。局部Schwartzman反应模型被用于本实验中。局部Schwartzman反应(LSR)是一种脂多糖(LPS)诱导的应答,该应答具有来自先天性免疫系统的细胞元件和体液元件的充分表征的作用。LSR对补体的依赖是非常确定的(Polak,L.等人,Nature 223:738-739(1969);Fong J.S.等人,J Exp Med 134:642-655(1971))。在LSR模型中,用TNFα(500ng,阴囊内)预处理小鼠达4小时,然后使小鼠麻醉并且准备用于提睾肌的活体显微镜检查。选择具有良好血流(1-4mm/s)的毛细血管后微静脉(15-60μm直径)网用于观察。用荧光抗体处理动物以选择性标记嗜中性粒细胞或血小板。顺序扫描血管网,并且数字记录所有血管的图像用于后面的分析。在记录微循环的基本状况之后,小鼠接受了单一LPS(100μg)的静脉内注射,其单独注射或与下列药剂一同注射。然后每10分钟扫描同一血管网达1小时。荧光团的特异性累积通过本底荧光的扣除来识别,并且通过确定图像的阈值而增强。反应的量级根据记录的图像测定。Schwartzman反应的初步测定是凝集数据。
所述研究比较了暴露于已知的补体途径耗竭剂眼镜蛇毒因子(CVF)或终末途径抑制剂(C5aR拮抗剂)的MASP-2+/+充足或野生型小鼠。结果(图29A)证明,CVF以及C5aR拮抗剂两者均防止了脉管系统中凝集体的出现。另外,MASP-2-/-缺陷小鼠(图29B)也证明了局部Schwartzman反应的完全抑制,支持凝集素途径的牵涉。这些结果清楚地证明MASP-2在DIC发生中的作用,并且支持利用MASP-2抑制剂治疗和预防DIC。
实施例34
本实施例描述鼠心肌缺血/再灌注模型中的MASP-2(-/-)小鼠的分析。
背景/基本原理:
为了评定MASP-2对冠状动脉的缺血性损伤之后的炎性再灌注损害的作用,在如Marber等人,J.Clin Invest.95:1446-1456(1995)所述的鼠缺血/再灌注(MIRP)模型中和在Langendorff分离灌注小鼠心脏模型中比较了MASP-2(-/-)和MASP-2(+/+)小鼠。
方法:
如实施例27中所述产生用于本研究的MASP-2(-/-)小鼠。使用实施例27中所述的方法在八只WT(MASP-2(+/+))小鼠和十一只MASP-2(-/-)小鼠中实施左心室的缺血性损伤。如实施例27中所述,梗塞尺寸(INF)和危险区(AAR)由测面积法确定。
Langendorff分离-灌注小鼠心脏模型:制备来自小鼠的心脏用于Langendorff分离-灌注小鼠心脏模型的方法按照F.J.Sutherland等人,Pharmacol Res 41:613(2000)中所述实施。也参见A.M.Kabir等人,Am J Physiol Heart Circ Physiol 291:H1893(2006);Y.Nishino等人,Circ Res 103:307(2008)和I.G.Webb等人,Cardiovasc Res(2010)。
简要描述,用戊巴比妥(300mg/kg)和肝素(150单位)腹膜内麻醉六只雄性WT(+/+)小鼠和九只雄性MASP-2(-/-)小鼠。心脏被迅速分离并且放在冰冷的改性Krebs-Henselit缓冲液(KH,118.5mmol/l NaCl、25.0mmol/l NaHCO3、4.75mmol KCl、KH2PO4 1.18、MgSO41.19、D-葡萄糖 11.0以及CaCl2 1.41)中。将切除的心脏安装到具有水套的Langendorff设备上,并且用95%O2和5%CO2平衡的KH缓冲液在80mm Hg的恒压下逆行灌注。灌注液的温度维持在37℃。插入左心室的充满流体的球囊监测了可收缩功能。逐渐使该球囊膨胀,直到舒张末期压在1mm Hg到7mm Hg。用0.075mm银丝(Advent)以580bpm进行心房起搏。冠脉血流通过灌注液的定时收集来测定。
体外梗塞评定
在逆行灌注开始之后,使心脏稳定达30分钟。总括而言,所有心脏不得不满足以下标准:冠脉血流量在1.5至4.5mL/min,心率>300bpm(非起搏),左心室形成压>55mm Hg,从开胸术到主动脉插管的时间<3min,并且在稳定化期间没有持续的节律障碍。然后在无血清的情况下实施全心缺血和再灌注。然后所有心脏通过夹住主动脉流入管经受30分钟的全心缺血,继之以2小时的再灌注。
电起搏在缺血期间收缩停止时终止,并且重新启动30分钟进入再灌注。在2小时的再灌注之后,用5ml溶于KH的1%氯化三苯基四唑(triphenyl tetrazolium chloride;TTC)灌注心脏达1分钟,并且然后在37℃下将心脏放在相同的溶液中达10分钟。然后除去心房,并且将心脏吸干、称重,并且-20℃储存达至多1周。
然后将心脏解冻,放在2.5%戊二醛中达1分钟,并且置入5%琼脂糖中。然后使用振动切片机(Agar Scientific)以0.7mm切片从顶端向底座切断琼脂糖心脏块。切片后,在4℃下将切片转移至PBS中达外加的一天以前,在室温下将切片放在10%甲醛中过夜。然后切片在Perspex板(相隔0.57mm)之间压缩并且使用扫描仪(Epson model G850A)成像。放大之后,使用图像分析软件(SigmaScan Pro 5.0,SPSS)和整体的表面积进行测面积法,并且TTC阴性的左心室心肌通过与组织厚度相乘而变换为体积。在每一心脏内,在个体切片的求和之后,将TTC阴性的梗塞体积表示为左心室容积的百分比或对左心室容积作图。
结果:
梗塞区域的尺寸(苍白),危险左心室(LV)区(红色)和正常灌注的LV区带(蓝色)通过它们的颜色外观和颜色边界的鉴定而在各个切片中描出轮廓。区域在每一切片的两面上定量并且由研究者进行平均。将梗塞体积计算为各个动物的风险区带的%(%RZ)。
图31A示出八只WT(+/+)小鼠和十一只MASP-2(-/-)小鼠经历上述冠状动脉闭塞和再灌注技术之后,对它们的梗塞尺寸的测定的评估。图31A将平均危险区(AAR,受缺血影响的区域的度量)和梗塞体积(INF,对心肌的损害的度量)图示为总心肌体积的百分比。如图31A中所示,尽管两个组之间的AAR没有差异,但MASP-2(-/-)小鼠中的INF体积与它们的WT同窝相比显著减少,因而指示在MIRP的这个模型中没有MASP-2的情况下免于心肌损伤的保护效应。
图31B将INF对AAR作图之间的关系图示为左心室(LV)心肌体积的%。如图31B中所示,对于任何给定的AAR,MASP-2(-/-)动物与它们的WT同窝相比显示它们的梗塞尺寸的高度显著的减少。
图31C和31D示出根据Langendorff分离-灌注小鼠心脏模型制备的WT(+/+)和MASP-2(-/-)小鼠的缓冲液灌注心脏的心肌梗塞的结果,其中全心缺血和再灌注在无血清的情况下实施。如图31C和图31D中所示,在MASP-2(-/-)小鼠的心脏与WT(+/+)小鼠的心脏之间没有观察到所得梗塞体积(INF)的差异,暗示图31A和图31B中所示的梗塞尺寸的差异是由血浆因子引起,而不是由MASP-2(-/-)小鼠的心肌组织对缺血性损害的较低易感性引起。
总之,这些结果证明,MASP-2缺乏在鼠心肌缺血/再灌注模型中缺血心脏的再灌注之后显著降低心肌损伤,并且支持利用MASP-2抑制剂治疗和预防缺血/再灌注损伤。
实施例35
本实施例描述鼠肾移植模型中的MASP-2(-/-)小鼠的分析。
背景/基本原理:
利用小鼠模型评定MASP-2在肾移植的功能性后果中的作用。
方法:
利用将单肾同基因移植至单侧肾切除后的受体小鼠中评定肾移植的功能性后果,其中使用六只WT(+/+)移植受体(B6)和六只MASP-2(-/-)移植受体。为了评定移植肾的功能,在移植后第5天从受体除去残存的天然肾,并且通过血尿素氮(BUN)水平的测定在24小时之后评定肾功能。
结果:
图32图示WT(+/+)受体和MASP-2(-/-)受体中肾移植后第6天的肾的血尿素氮(BUN)水平。如图32中所示,在WT(+/+)(B6)移植受体内观察到大大升高的BUN水平(小鼠中的正常BUN水平<5mM),指示肾衰竭。相反,MASP-2(-/-)同基因移植受体小鼠显示显著较低的BUN水平,暗示改善的肾功能。已注意到,这些结果是利用来自WT(+/+)肾供体的移植物而获得的,暗示仅仅移植受体中的功能性凝集素途径的缺少就足以实现治疗益处。
总之,这些结果表明,经由MASP-2抑制的凝集素途径的瞬时抑制在肾移植中提供降低发病率和延迟的移植物功能的方法,并且此方法可能适用于其他移植情况。
实施例36
本实施例证明,MASP-2(-/-)小鼠在鼠多微生物败血症腹膜炎模型中是抗败血症休克的。
背景/基本原理:
为了评估MASP-2(-/-)在感染中的潜在作用,评估了盲肠结扎和穿刺(CLP)模型,其为一种多微生物败血症腹膜炎的模型。这个模型被认为最精确地模拟人败血症腹膜炎的过程。盲肠结扎和穿刺(CLP)模型是其中盲肠由针结扎和穿刺、导致细菌连续渗漏进入腹腔、细菌经由淋巴引流到达血液并且然后分布至所有腹器官中、导致多器官衰竭和败血症休克的模型(Eskandari等人,J Immunol 148(9):2724-2730(1992))。CLP模型模拟患者中观察到的败血症的过程,并且诱发早期高炎性应答,继之以明显的低炎性阶段。在这个阶段期间,动物对细菌激发高度敏感(Wichterman等人,J.Surg.Res.29(2):189-201(1980))。
方法:
如实施例23中所述在WT(+/+)(n=18)和MASP-2(-/-)(n=16)小鼠中测定利用盲肠结扎和穿刺(CLP)模型的多微生物感染的死亡率。简述之,将MASP-2缺陷小鼠和它们的野生型同窝麻醉,并且取出盲肠并且在远端上方30%处结扎。其后,用0.4mm直径的针穿刺盲肠一次。然后将盲肠放到腹腔中,并且用夹具闭合皮肤。在CLP后14天的时期内监测经受了CLP的小鼠的存活。CLP后16小时在小鼠中收集腹膜灌洗液以测定细菌负载。腹膜灌洗液的连续稀释物在PBS中制备并且在Mueller Hinton板中接种,随后在厌氧条件下37℃温育达24小时,之后测定细菌负载。
在肺和脾中CLP之后16小时,对细菌感染的TNF-α细胞因子应答也在WT(+/+)和MASP-2(-/-)小鼠中经由定量实时聚合酶链式反应(qRT-PCR)测定。在WT(+/+)和MASP-2(-/-)小鼠中CLP之后16小时,TNF-α的血清水平也通过夹心ELISA定量。
结果:
图33将CLP处理的动物的存活百分比图示为CLP操作之后天数的函数。如图33中所示,与WT(+/+)小鼠相比,MASP-2(-/-)小鼠中的凝集素途径缺乏不增加利用盲肠结扎和穿刺模型的多微生物感染后的小鼠的死亡率。然而,如图34中所示,与它们的WT(+/+)同窝相比时,MASP-2(-/-)小鼠显示了CLP之后腹膜灌洗液中显著更高的细菌负载(细菌数增加大约1000倍)。这些结果表明,MASP-2(-/-)缺陷小鼠是抗败血症休克的。此模型中的MASP-2缺陷小鼠中减少的细菌清除可能归因于削弱的C3b介导的吞噬作用,因为证明了C3沉积是MASP-2依赖性的。
已测定,与WT(+/+)对照相比,MASP-2(-/-)小鼠中的对细菌感染的TNF-α细胞因子应答没有升高(未显示数据)。同样已测定,与TNF-α的血清水平保持几乎不变的MASP-2(-/-)小鼠相反,在CLP后的16小时,WT(+/+)小鼠中存在显著更高的TNF-α的血清浓度。这些结果暗示,对败血症状况的强烈炎性应答在MASP-2(-/-)小鼠中得到缓和,并且允许动物在更高细菌计数的情况下存活。
总之,这些结果证明就败血症而言凝集素途径补体活化的潜在有害效应和压倒性败血症的患者中增加的死亡率。这些结果进一步证明,MASP-2缺乏调节炎性免疫反应并且降低败血症期间炎性介质的表达水平。因此认为,通过给予对抗MASP-2的抑制性单克隆抗体抑制MASP-2(-/-)将有效减少患有败血症休克的受试者中的炎性应答。
实施例37
本实施例描述鼠鼻内感染性模型中MASP-2(-/-)小鼠的分析。
背景/基本原理:
绿脓假单胞菌(Pseudomonas aeruginosa)是一种革兰氏阴性机会性人细菌性病原体,该病原体引起大范围的感染,特别是在免疫受损的个体中。它是获得性医院感染的主要来源,特别是医院获得性肺炎。它也在囊性纤维化(CF)患者中造成显著的发病率和死亡率。绿脓假单胞菌肺感染的特征在于强的嗜中性粒细胞募集和导致广泛组织损伤的显著的肺脏炎症(PalankiM.S.等人,J.Med.Chem 51:1546-1559(2008))。
在本实施例中,进行研究以确定MASP-2(-/-)小鼠中的凝集素途径的去除是否增加小鼠对细菌感染的易感性。
方法:
用绿脓假单胞菌菌株的鼻内给药激发22只WT(+/+)小鼠、22只MASP-2(-/-)小鼠和11只C3(-/-)小鼠。所述小鼠在感染后受监测超过六天,并且构建Kaplan-Mayer曲线,显示存活百分比。
结果:
图35是感染后六天WT(+/+)、MASP-2(-/-)或C3(-/-)小鼠的存活百分比的Kaplan-Mayer曲线。如图35中所示,与WT(+/+)小鼠比较,在MASP-2(-/-)小鼠中未观察到差异。然而,C3(-/-)小鼠中经典(C1q)途径的去除导致对细菌感染的严重的易感性。这些结果证明,MASP-2抑制不增加对细菌感染的易感性,表明有可能通过抑制MASP-2来减少外伤患者中不希望的炎性并发症而不减损患者利用经典补体途径抗感染的能力。
实施例38
本实施例描述如实施例24中所述鉴定的代表性高亲和力抗MASP-2Fab2抗体的药效分析。
背景/基本原理:
如实施例24中所述,为了识别封闭大鼠凝集素途径的高亲和力抗体,利用大鼠MASP-2蛋白淘选噬菌体展示文库。这个文库经设计以提供高免疫多样性并且完全利用人类免疫球蛋白基因序列而构建。如实施例24中所示,通过ELISA筛选识别了大约250个独立的噬菌体克隆,所述噬菌体克隆以高亲和力与大鼠MASP-2蛋白结合。这些克隆的测序鉴定了50个独特的MASP-2抗体编码噬菌体。由这些克隆表达Fab2蛋白,纯化和分析Fab2蛋白的MASP-2结合亲和力和凝集素补体途径功能性抑制。
如实施例24的表6中所示,具有功能性封闭活性的17个抗MASP-2Fab2被鉴定为此分析的结果(对于封闭性抗体而言,34%命中率)。凝集素补体途径通过Fab2的功能性抑制在C4沉积的水平上是明显的,C4沉积的水平是由MASP-2进行的C4裂解的直接度量。重要地,当评定C3转化酶活性时,抑制同样明显,证明凝集素补体途径的功能性阻断。如实施例24中所述识别的所述17个MASP-2封闭性Fab2有力地抑制C3转化酶形成,其IC50值等于或小于10nM。所识别的17个Fab2中的八个具有亚纳摩尔范围内的IC50值。此外,所有17个MASP-2封闭性Fab2在凝集素途径C3转化酶试验中给出C3转化酶形成的基本完全的抑制,如图11A至图11C中所示,并且概括在实施例24的表6中。此外,表6中所示的17个封闭性抗MASP-2Fab2中的每一个有力地抑制C3b生成(>95%),因而证明本试验针对凝集素途径C3转化酶的特异性。
大鼠IgG2c和小鼠IgG2a全长抗体同种型变体来源于Fab2#11。本实施例描述这些同种型对于药效参数的体内表征。
方法:
如实施例24中所述,大鼠MASP-2蛋白用于淘选Fab噬菌体展示文库,Fab2#11是从该文库中鉴定的。大鼠IgG2c和小鼠IgG2a全长抗体同种型变体来源于Fab2#11。大鼠IgG2c和小鼠IgG2a全长抗体同种型均如下在体内对于药效参数进行表征。
小鼠中的体内研究:
在小鼠中进行药效研究,以研究抗MASP-2抗体给药对体内血浆凝集素途径活性的作用。在此研究中,在凝集素途径试验中,在小鼠抗MASP-2MoAb(来源于Fab2#11的小鼠IgG2a全长抗体同种型)的0.3mg/kg或1.0mg/kg的皮下(sc)和腹膜内(ip)给药之后的不同时间点离体测定C4沉积。
图36图示凝集素途径特异性C4b沉积,其在取自小鼠(n=3小鼠/组)的未稀释的血清样品中、在小鼠抗MASP-2MoAb的0.3mg/kg或1.0mg/kg皮下给药之后的不同时间点离体测定。在抗体给药之前收集的来自小鼠的血清样品充当阴性对照(100%活性),而体外补充了100nM的同样的封闭性抗MASP-2抗体的血清用作阳性对照(0%活性)。
图36中所示的结果证明在小鼠抗MASP-2MoAb的1.0mg/kg剂量皮下给药之后的C4b沉积的快速和完全的抑制。在小鼠抗MASP-2MoAb的0.3mg/kg剂量的皮下给药之后,观察到C4b沉积的部分抑制。
小鼠抗MASP-2MoAb在小鼠中以0.6mg/kg单次腹膜内给药之后,跟踪凝集素途径恢复的时间过程达三个星期。如图37中所示,凝集素途径活性的急剧下降发生在抗体给药之后,继之以腹膜内给药后持续约7天的完全的凝集素途径抑制。经过第二和第三周,观察到凝集素途径活性的缓慢恢复,小鼠中完全的凝集素途径恢复则是在抗MASP-2MoAb给药后17天。
这些结果证明,来源于Fab2#11的小鼠抗MASP-2MoAb在全身输送时以剂量响应的方式抑制小鼠的凝集素途径。
实施例39
本实施例描述对来源于Fab2#11的小鼠抗MASP-2MoAb在年龄相关黄斑变性小鼠模型中效力的分析。
背景/基本原理:
如实施例24中所述,大鼠MASP-2蛋白用于淘选Fab噬菌体展示文库,Fab2#11从该文库中鉴定为功能活性抗体。大鼠IgG2c和小鼠IgG2a同种型的全长抗体由Fab2#11产生。如实施例38中所述表征了小鼠IgG2a同种型的全长抗MASP-2抗体的药效参数。在本实施例中,在年龄相关黄斑变性(AMD)的小鼠模型中分析了来源于Fab2#11的小鼠抗MASP-2全长抗体,该小鼠模型由Bora P.S.等人,J Immunol 174:491-497(2005)描述。
方法:
如中实施例38所述的来源于Fab2#11的小鼠IgG2a全长抗MASP-2抗体同种型在年龄相关黄斑变性(AMD)的小鼠模型中测试,如实施例28中所述并具有以下修改。
小鼠抗MASP-2MoAb的给药
将两种不同剂量(0.3mg/kg和1.0mg/kg)的小鼠抗MASP-2MoAb连同同种型对照MoAb处理一起在CNV诱发之前16小时腹膜内注射至WT(+/+)小鼠(n=8小鼠/组)。
脉络膜新生血管形成(CNV)的诱发:
利用如实施例28中所述的激光凝固进行脉络膜新生血管形成(CNV)的诱发和CNV体积的测定。
结果:
图38图示在用同种型对照MoAb或小鼠抗MASP-2MoAb(0.3mg/kg和1.0mg/kg)处理的小鼠中激光损伤后7天测定的CNV面积。如图38中所示,在用1.0mg/kg抗MASP-2MoAb预处理的小鼠中,激光处理后七天观察到CNV的统计显著的(p<0.01)大约50%减少。如图38中进一步示出,观察到0.3mg/kg剂量的抗MASP-2MoAb对减少CNV并不有效。注意到,0.3mg/kg剂量的抗MASP-2MoAb显示出在皮下给药之后具有C4b沉积的部分和瞬时抑制,如实施例38中所述和图36中所示。
本实施例中所述的结果证明,利用抑制剂(诸如抗MASP-2MoAb)的MASP-2的阻断在黄斑变性的治疗中具有预防和/或治疗作用。注意到,这些结果与实施例28中所述的在MASP-2(-/-)小鼠中进行的研究中观察到的结果一致,其中,与野生型对照小鼠相比,在MASP-2(-/-)小鼠中观察到激光处理后7天的CNV的30%减少。此外,本实施例中的结果进一步证明,全身输送的抗MASP-2抗体在眼内提供局部的治疗益处,从而强调全身性给药途径治疗AMD患者的潜力。概括而言,这些结果提供支持在AMD的治疗中利用MASP-2MoAb的证据。
实施例40
本实施例证明,与野生型对照小鼠相比,MASP-2缺陷小鼠受到保护而免于脑膜炎奈瑟氏菌感染后的脑膜炎奈瑟氏菌诱发的死亡率,并且具有增强的菌血症清除。
基本原理:脑膜炎奈瑟氏菌是异养革兰氏阴性双球菌,它在脑膜炎及其他形式的脑膜炎球菌疾病(诸如脑膜炎球菌血症)中的作用是已知的。脑膜炎奈瑟氏菌是儿童期间的致病率和死亡率的主要诱因。严重的并发症包括败血症、华-弗综合征Waterhouse-Friderichsen syndrome)、肾上腺机能不全和弥散性血管内凝血(DIC)。见例如Rintala E.等人,Critical Care Medicine28(7):2373-2378(2000)。在本实施例中,在MASP-2(-/-)和WT(+/+)小鼠中分析凝集素途径的作用,以便解决MASP-2缺陷小鼠是否会对脑膜炎奈瑟氏菌诱发的死亡率敏感。
方法:
如实施例27中所述产生MASP-2敲除小鼠。10周大的MASP-2KO小鼠(n=10)和野生型C57/B6小鼠(n=10)通过静脉内注射被接种一剂5×108cfu/100μl、2×108cfu/100μl或3×107cfu/100μl的溶于400mg/kg右旋糖酐铁的脑膜炎奈瑟氏菌血清群A Z2491。监测感染后的小鼠的存活超过72小时时间周期。感染后,以每小时一次的间隔从小鼠采集血样,并且分析所述血样以测定脑膜炎奈瑟氏菌的血清水平(log cfu/ml),以便验证感染并且测定细菌从血清的清除率。
结果:
图39A图示在5×108/100μl cfu感染剂量的脑膜炎奈瑟氏菌给药之后,MASP-2KO小鼠和WT小鼠的存活百分比。如图39A中所示,在用5×108/100μl cfu的最高剂量的脑膜炎奈瑟氏菌感染之后,100%的MASP-2KO小鼠在感染后的72小时期间自始至终存活。相反,仅20%的WT小鼠在感染后的24小时仍然活着。这些结果证明,MASP-2缺陷小鼠受到保护而免于脑膜炎奈瑟氏菌诱发的死亡率。
图39B图示取自用5×108cfu/100μl脑膜炎奈瑟氏菌感染的MASP-2KO小鼠和WT小鼠的血样中在不同时间点回收的脑膜炎奈瑟氏菌的log cfu/ml。如图39B中所示,在WT小鼠中,血液中的脑膜炎奈瑟氏菌水平在感染后24小时达到约6.5log cfu/ml的峰值,并且在感染后48小时下降到零。相反,在MASP-2KO小鼠中,脑膜炎奈瑟氏菌水平在感染后6小时达到约3.5log cfu/ml的峰值,并且在感染后36小时下降到零。
图40A图示MASP-2KO小鼠和WT小鼠在由2×108cfu/100μl脑膜炎奈瑟氏菌感染之后的存活百分比。如图40A中所示,在用2×108cfu/100μl剂量的脑膜炎奈瑟氏菌感染之后,100%的MASP-2KO小鼠在感染后的72小时期间自始至终存活。相反,仅80%的WT小鼠在感染后24小时仍然活着。与图39A中所示的结果一致,这些结果进一步证明,MASP-2缺陷小鼠受到保护而免于脑膜炎奈瑟氏菌诱发的死亡率。
图40B图示取自用2×108cfu/100μl脑膜炎奈瑟氏菌感染的WT小鼠的血样中在不同时间点回收的脑膜炎奈瑟氏菌的log cfu/ml。如图40B中所示,用2×108cfu感染的WT小鼠的血液中的脑膜炎奈瑟氏菌水平在感染后12小时达到约4log cfu/ml的峰值,并且在感染后24小时下降到零。图40C图示取自用2×108cfu/100μl脑膜炎奈瑟氏菌感染的MASP-2KO小鼠的血样中在不同时间点回收的脑膜炎奈瑟氏菌的log cfu/ml。如图40C中所示,用2×108cfu感染的MASP-2KO小鼠的血液中的脑膜炎奈瑟氏菌水平在感染后2小时达到约3.5log cfu/ml的峰水平,并且在感染后3小时下降到零。与图39B中所示的结果一致,这些结果证明,尽管用与WT小鼠相同剂量的脑膜炎奈瑟氏菌感染MASP-2KO小鼠,但与WT相比,MASP-2KO小鼠具有增强的菌血症清除。
在用3×107cfu/100μl的最低剂量的脑膜炎奈瑟氏菌感染之后,MASP-2KO和WT小鼠的存活百分比在72小时时期处为100%(未显示数据)。
讨论
这些结果显示,与WT小鼠相比,MASP-2缺陷小鼠受到保护而免于脑膜炎奈瑟氏菌诱发的死亡率,并且具有增强的菌血症清除。因此,鉴于这些结果预期,MASP-2抑制剂(诸如MASP-2MoAb)的治疗应用将被期待以有效治疗、预防或减轻脑膜炎奈瑟氏菌感染的影响(即,败血症和DIC)。此外,这些结果表明,MASP-2抑制剂(诸如MASP-2MoAb)的应用不会将受试者预置于染上脑膜炎奈瑟氏菌感染的增大的风险中。
虽然描述并说明了优选的实施方案,但是应当理解的是,可以在不偏离本发明的精神和范围的情况下在其中进行各种修改。
- 还没有人留言评论。精彩留言会获得点赞!
- 干扰素调节因子5(IRF5)及其抑制剂在治疗心肌肥厚中的应用的制作方法与工艺
- 作为因子XIA抑制剂的环状P1连接体的制作方法与工艺
- 能够结合组织因子途径抑制剂(1‑161)上的两个表位的抗体的制作方法与工艺
- 针对组织因子途径抑制剂(TFPI)的优化的单克隆抗体的制作方法与工艺
- 一种作为自分泌运动因子抑制剂的咔唑类化合物及其制备方法和应用与流程
- 外周血单核细胞活化抑制剂在制备治疗急性肝衰竭的药物中的用途的制造方法与工艺
- 对NEDD8活化酶(NAE)抑制剂的反应的生物标记的制造方法与工艺
- 一种肺癌调节因子SRSF5及其抑制剂与应用的制造方法与工艺
- 自分泌运动因子抑制剂的制造方法与工艺
- 一种BCL‑2抑制剂Venetoclax的合成方法与流程