用于治疗疼痛、炎症、炎性疾病、免疫或自身免疫疾病和肿瘤生长的单独的或与类固醇组合的(R)-卡比多巴的制作方法
本发明涉及治疗或预防例如疼痛、炎性疾病、免疫或自身免疫病和肿瘤生长的医学病症的(r)-卡比多巴和包含它的药物组合物。本发明还涉及包含(r)-卡比多巴与类固醇的组合制剂,以及所述组合制剂在医学中(特别是在治疗或预防例如疼痛、炎性疾病、免疫或自身免疫病的医学病症以及抑制肿瘤生长中)的用途。
背景技术
氨基脲敏感性胺氧化酶(semicarbazide-sensitiveamineoxidase,ssao),也称为血管黏附蛋白-1(vascularadhesionprotein-1,vap-1)或含铜胺氧化酶3(amineoxidase,coppercontaining3,aoc3),属于酶的含铜胺氧化酶家族(ec.1.4.3.6)。该酶家族的成员对氨基脲抑制敏感,并且根据以下反应在伯胺至醛、过氧化氢和氨的氧化脱氨中利用铜离子和蛋白质来源的多巴醌(topaquinone,tpq)辅因子:
r-ch2-nh2+o2→r-cho+h2o2+nh3
人ssao的已知底物包括内源性甲胺和氨基丙酮以及一些异生胺(xenobioticamine)例如苄胺[lyles,int.j.biochem.cellbiol.1996,28,259-274;klinman,biochim.biophys.acta2003,1647(1-2),131-137;mátyus等,curr.med.chem.2004,11(10),1285-1298;o′sullivan等,neurotoxicology2004,25(1-2),303-315]。与其他含铜胺氧化酶类似,dna序列分析和结构确定表明组织结合的人ssao是由通过单一n端跨膜结构域锚定至质膜的两个90至100kda亚基组成的同二聚体糖蛋白[morris等,j.biol.chem.1997,272,9388-9392;smith等,j.exp.med.1998,188,17-27;airenne等,proteinscience2005,14,1964-1974;jakobsson等,actacrystallogr.dbiol.crystallogr.2005,61(pt11),1550-1562]。
已经在多种组织(包括血管和非血管平滑肌组织、内皮、以及脂肪组织)中发现ssao活性[lewinsohn,braz.j.med.biol.res.1984,17,223-256;nakos和gossrau,foliahistochem.cytobiol.1994,32,3-10;yu等,biochem.pharmacol.1994,47,1055-1059;castillo等,neurochem.iht.1998,33,415-423;lyles和pino,j.neural.transm.suppl.1998,52,239-250;jaakkola等,am.j.pathol.1999,155,1953-1965;morin等,j.pharmacol.exp.ther.2001,297,563-572;salmi和jalkanen,trendsimmunol.2001,22,211-216]。此外,在血浆中发现ssao蛋白并且该可溶性形式显示具有与组织结合形式相似的特性[yu等,biochem.pharmacol.1994,47,1055-1059;
这种丰富的酶的精确生理学作用尚未完全确定,但是显示ssao及其反应产物在细胞信号传导和调节中可具有数种功能。例如,最近的发现表明ssao在glut4介导的葡萄糖摄取[enrique-tarancon等,j.biol.chem.1998,273,8025-8032;morin等,j.pharmacol.exp.ther.2001,297,563-572]和脂肪细胞分化[fontana等,biochem.j.2001,356,769-777;mercier等,biochem.j.2001,358,335-342]中发挥作用。此外,已经表明ssao作为白细胞的黏附蛋白参与炎性过程[salmi和jalkanen,trendsimmunol.2001,22,211-216;salmi和jalkanen,在“adhesionmolecules:functionsandinhibition”中k.ley(编辑),2007,第237-251页],并且还可在结缔组织基质发育和维持中发挥作用[langford等,cardiovasc.toxicol.2002,2(2),141-150;
在人中的数个研究表明在例如充血性心力衰竭、糖尿病、阿尔茨海默病(alzheimer′sdisease)和炎症的病症中,血浆中的ssao活性提高[lewinsohn,braz.j.med.biol.res.1984,17,223-256;boomsma等,cardiovasc.res.1997,33,387-391;ekblom,pharmacol.res.1998,37,87-92;
wo2007/146188教导阻断ssao活性抑制白细胞招募、降低炎性应答,并且预期其在预防和治疗例如癫痫的发作中是有益的。
o′rourke等(jneuraltransm.2007;114(6):845-9)检查了ssao抑制剂在神经疾病中的潜力,先前已表明在卒中的大鼠模型中ssao抑制的效力。在复发缓解型实验性自身免疫性脑脊髓炎(experimentalautoimmuneencephalomyelitis,eae)(与人多发性硬化共有许多特征的小鼠模型)上测试ssao抑制剂。数据示出在该模型中并因此在治疗人多发性硬化中小分子抗ssao治疗的潜在临床益处。
ssao敲除动物在表型上明显正常,但是其在响应于多种炎性刺激诱发的炎性应答中表现出显著的降低[stolen等,immunity2005,22(1),105-115]。此外,通过使用抗体和/或小分子,在多种人疾病的动物模型(例如,角叉聚糖诱导的爪炎症、
当炎症的消退部分地被炎性刺激的慢性性质消除时,慢性组织炎症可导致纤维化。结果可以是具有过度细胞外基质沉积(包括胶原蛋白)和组织疤痕的组织的不合适修复。这是导致胶原蛋白、弹性蛋白、透明质酸、糖蛋白和蛋白聚糖产生提高的包括纤连蛋白和活性氧类(reactiveoxygenspecies)以及生长因子(例如转化生长因子β-1(transforminggrowthfactor-β-1,tgfβ-1)、胰岛素样生长因子-i(insulin-likegrowthfactor-i,igf-i)、血小板源性生长因子(platelet-derivedgrowthfactor,pdgf)和结缔组织生长因子(connectivetissuegrowthfactor,ctgf))的刺激的肌成纤维细胞活化的结果。此外,侵入的巨噬细胞的活性在调节修复和纤维化过程中发挥关键作用。
vap-1也参与尤其是在肝中的纤维化疾病的进展和维持。weston和adams(jneuraltransm.2011,118(7),1055-64)已总结了涉及肝纤维化中的vap-1的实验数据。weston等(easlposter2010)示出人纤维化肝中高度提高的vap-1表达,特别是与活化的肌成纤维细胞和胶原原纤维相关。这一与纤维化的解剖学关联与vap-1的阻断加速了四氯化碳诱导性纤维化的消退的观察结果一致,并且表明vap-1/ssao酶产物h2o2在肌成纤维细胞的活化中的作用。同一作者还示出促纤维化生长因子tgfβ使肝细胞中vap-1的表达提高约50倍。此外,vap-1参与肺炎症(例如singh等,2003,virchowsarch442:491-495),表明vap-1阻断剂会减轻肺炎症,并因此对通过治疗疾病的促纤维化和促炎方面二者来治疗囊性纤维化有益处。
ssao(vap-1)在胃癌中上调并且已经在人黑素瘤、肝细胞肿瘤(hepatoma)和头颈部肿瘤的肿瘤脉管系统中鉴定(yoongkf,mcnabg,hubschersg,adamsdh.(1998),jimmunol160,3978-88.;irjalah,salmim,alanenk,gre′nmanr,jalkanens(2001),immunol.166,6937-6943;forster-horvathc,domeb,pakus等(2004),melanomares.14,135-40.)。一份报道(marttila-ichiharaf,castermansk,auvinenk,oudeegbrinkmg,jalkanens,griffioenaw,salmim.(2010),jimmunol.184,3164-3173.)已经显示具有酶失活的vap-1的小鼠生长黑素瘤更慢,并且具有降低的肿瘤血管数量和直径。这些肿瘤生长的降低也反映在髓样抑制细胞的降低(60%至70%)的浸润中。鼓舞人心地,vap-1缺陷对正常组织中血管或淋巴形成没有影响。
由于以上原因,预期ssao的抑制将降低促炎酶产物(醛、过氧化氢和氨)的水平,同时也降低免疫细胞的黏附能力并相应地降低其活化和最终的外渗。预期其中这样的活性在治疗上有益的疾病包括在其中免疫细胞在病理状况的起始、维持或消退中发挥显著作用的所有疾病,例如炎性疾病和免疫/自身免疫病。此类疾病的实例包括多发性硬化、关节炎和血管炎。
发明概述
本申请人出乎意料地发现(r)-卡比多巴具有可用的ssao/vap-1抑制活性,并且(r)-卡比多巴可用于治疗或预防其中vap-1活性的抑制具有益处的医学病症,例如炎性疾病、免疫或自身免疫病和肿瘤生长。
此外,本申请人出乎意料地发现(r)-卡比多巴在治疗疼痛(包括炎性疼痛)中有效。
此外,本申请人已发现(r)-卡比多巴对于ssao/vap-1具有超过酶多巴脱羧酶的出乎意料的选择性。这是尤其出乎意料的,因为包含(s)-卡比多巴的药物
此外,本申请人已使得可获得(r)-卡比多巴与类固醇的组合制剂。预期该组合制剂可用于治疗或预防在其中抑制vap-1活性具有益处的医学病症,例如炎性疾病、免疫或自身免疫病和肿瘤生长。
此外,本申请人已发现(r)-卡比多巴与类固醇的组合制剂对于治疗疼痛(包括炎性疼痛)出乎意料地有效。
附图简述
以下参照附图描述本发明的一些实施方案,其中:
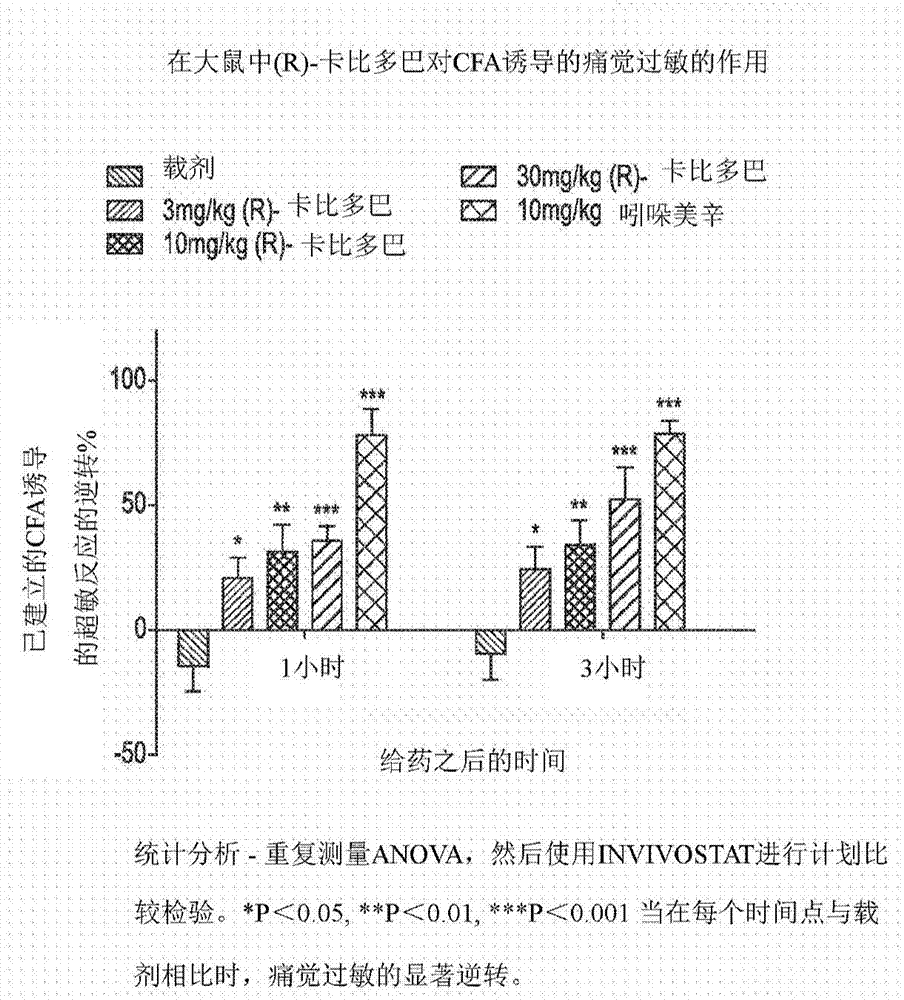
图1示出了给药之后1小时和3小时在大鼠中(r)-卡比多巴对cfa诱导的痛觉过敏的作用(左至右-载剂;3mg/kg(r)-卡比多巴;10mg/kg(r)-卡比多巴;30mg/kg(r)-卡比多巴;10mg/kg吲哚美辛);
图2示出了给药之后3小时在大鼠中(r)-卡比多巴和吲哚美辛对cfa诱导的痛觉过敏中爪水肿的作用(左至右-载剂/载剂;3mg/kg(r)-卡比多巴/载剂;10mg/kg(r)-卡比多巴/载剂;30mg/kg(r)-卡比多巴/载剂;10mg/kg吲哚美辛);
图3示出了给药之后1小时和3小时在大鼠中泼尼松龙对cfa诱导的痛觉过敏的作用(左至右-载剂/载剂;0.3mg/kg泼尼松龙/载剂;1mg/kg泼尼松龙/载剂;3mg/kg泼尼松龙/载剂;10mg/kg泼尼松龙/载剂;10mg/kg吲哚美辛);以及
图4示出了给药之后1小时和3小时在大鼠中(r)-卡比多巴和泼尼松龙对cfa诱导的痛觉过敏的作用(左至右-载剂/载剂;3mg/kg(r)-卡比多巴/载剂;10mg/kg(r)-卡比多巴/载剂;0.3mg/kg泼尼松龙/载剂;3mg/kg(r)-卡比多巴/0.3mg/kg泼尼松龙,10mg/kg(r)-卡比多巴/0.3mg/kg泼尼松龙)。
发明详述
定义
本文中使用的术语“治疗”是指获得期望的药理学和/或生理学作用。例如,在治疗疼痛的情况下,所述作用在完全地或部分地预防疼痛或其症状方面可以是预防性的,和/或在疼痛的部分或完全治愈和/或由于疾病引起的不良作用方面可以是治疗性的。本文中使用的“治疗”包括在哺乳动物中(特别是在人中)的任何疼痛治疗,并且包括(a)在易感疾病但尚未诊断出患有该疾病的对象中预防疾病发生;(b)抑制该疾病,即阻止其发生;以及(c)减轻疾病,即,使疾病的消退。
(r)-卡比多巴和/或类固醇的“有效量”是指当向哺乳动物或其他对象施用用于治疗疾病或病症时足以对所述疾病或病症产生这样的治疗的(r)-卡比多巴和/或类固醇的量。“有效量”将根据类固醇(如果有的话),疾病及其严重程度,以及待治疗的对象的年龄、体重等而改变。治疗效果可以是客观的(即,可通过一些测试或标志物测量)或主观的(即,对象给出对作用的指示或感觉到作用)。
“可药用”意指可用于制备通常安全、无毒且既不是生物学上也不是其他方面不期望的药物组合物,并且包括可用于兽用以及人药用。合适的可药用盐包括例如来源于无机酸或有机酸的酸加成盐,例如盐酸盐、氢溴酸盐、对甲苯磺酸盐、磷酸盐、硫酸盐、高氯酸盐、乙酸盐、三氟乙酸盐、丙酸盐、柠檬酸盐、丙二酸盐、琥珀酸盐、乳酸盐、草酸盐、酒石酸盐和苯甲酸盐。对于盐的综述,参见stahl和wermuth的handbookofpharmaceuticalsalts:properties,selection,anduse(wiley-vch,weinheim,germany,2002)。可药用盐也可用碱来形成。这样的盐包括来源于无机碱或有机碱的盐,例如碱金属盐(例如镁或钙的盐),以及有机胺盐(例如吗啉、哌啶、二甲胺或二乙胺的盐)。
本文中使用的术语“疼痛”包括炎性疼痛。在一个实施方案中,所述疼痛是炎性疼痛。
卡比多巴
卡比多巴以(r)和(s)对映体的形式存在。卡比多巴通常可作为(r)和(s)对映体的混合物获得。本文中提及的“(r)-卡比多巴”包括含有(r)卡比多巴的任何组合物或混合物,包括例如基本上纯的(r)-卡比多巴或(r)-卡比多巴和(s)-卡比多巴的混合物,例如外消旋混合物。
在本发明的实践中,可期望利用主要是(r)-卡比多巴的混合物,例如至少60%、至少70%、至少80%、至少90%或至少99%对映纯的(r)-卡比多巴的组合物。为了利用上述酶抑制选择性的益处,可期望使用富含(r)-卡比多巴的组合物。
类固醇
本文中使用的术语“类固醇”意指适用于根据本发明的组合制剂的任何类固醇。术语“类固醇”还旨在涵盖在本发明的组合物中和在实践本发明的方法中使用的两种或更多种类固醇的组合。
合适的类固醇包括糖皮质激素。糖皮质激素类固醇的一些实例包括泼尼松龙、泼尼松、甲泼尼龙、曲安西龙、地塞米松、氢化可的松、地夫可特(deflazacourt)、倍他米松和布地奈德,或其可药用盐。特别优选的类固醇包括泼尼松龙,或其可药用盐;以及泼尼松,或其可药用盐。
(r)-卡比多巴用于治疗疼痛
本申请人发现(r)-卡比多巴在治疗疼痛(包括炎性疼痛)中出乎意料地有效。本文中提供了表明在公认的疼痛模型中的效力的体内数据。参见例如图1。
此外,本申请人发现(r)-卡比多巴在治疗炎症中出乎意料地有效。本文中提供了表明在公认的炎症模型中的效力的体内数据。参见例如图2。
本发明的治疗可提供以下的任一项或所有:优异的疼痛或炎症降低;更快地减轻疼痛或炎症;提高的顺应性;降低的成瘾可能性;降低的治疗相关副作用;降低暴露于表现出剂量依赖性治疗相关副作用的其他治疗剂的能力;或任何其他可感知的治疗益处。
(r)-卡比多巴与类固醇的组合制剂
本申请人还发现与类固醇组合的(r)-卡比多巴在治疗疼痛中出乎意料地有效。出乎意料地有效意指(r)-卡比多巴和类固醇一起产生了大于当单独给药时(r)-卡比多巴和类固醇的治疗效果的治疗效果。在一个实施方案中,与类固醇组合的(r)-卡比多巴在治疗疼痛中提供了协同有益效果。
因此,在一个实施方案中,本发明使得可获得包含(r)-卡比多巴或者其水合物或可药用盐和类固醇或其可药用盐的组合制剂。在一个实施方案中,所述类固醇是糖皮质激素。在一个实施方案中,所述类固醇是选自以下的任一种的糖皮质激素:泼尼松龙、泼尼松、甲泼尼龙、曲安西龙、地塞米松、氢化可的松、地夫可特、倍他米松和布地奈德,或其可药用盐。在另一个实施方案中,所述类固醇是两种或更多种任何前述类固醇或其盐的组合。在一些特别的实施方案中,所述类固醇是泼尼松龙,或其可药用盐。在一些特别的实施方案中,所述类固醇是泼尼松,或其可药用盐。
本发明的组合可提供以下的任一项或所有:优异的疼痛或炎症降低;更快地减轻疼痛或炎症;提高的顺应性;降低的成瘾可能性;降低的治疗相关副作用;降低暴露于表现出剂量依赖性治疗相关副作用的其他治疗剂的能力;或任何其他可感知的治疗益处。
在一个实施方案中,所述组合制剂包含(r)-卡比多巴或者其水合物或可药用盐,以及泼尼松龙或其可药用盐。
在一个实施方案中,所述组合制剂包含(r)-卡比多巴或者其水合物或可药用盐,以及泼尼松或其可药用盐。
在一个实施方案中,所述组合制剂包含(r)-卡比多巴或者其水合物或可药用盐,以及甲泼尼龙或其可药用盐。
在一个实施方案中,所述组合制剂包含(r)-卡比多巴或者其水合物或可药用盐,以及曲安西龙或其可药用盐。
在一个实施方案中,所述组合制剂包含(r)-卡比多巴或者其水合物或可药用盐,以及地塞米松或其可药用盐。
在一个实施方案中,所述组合制剂包含(r)-卡比多巴或者其水合物或可药用盐,以及氢化可的松或其可药用盐。
在一个实施方案中,所述组合制剂包含(r)-卡比多巴或者其水合物或可药用盐,以及地夫可特或其可药用盐。
在一个实施方案中,所述组合制剂包含(r)-卡比多巴或者其水合物或可药用盐,以及倍他米松或其可药用盐。
在一个实施方案中,所述组合制剂包含(r)-卡比多巴或者其水合物或可药用盐,以及布地奈德或其可药用盐。
组合物
含有活性成分,或在组合制剂的情况下含有多种活性成分的药物组合物可以是任何合适的形式,例如水性或非水性溶液剂或混悬剂、可分散的散剂或颗粒剂、经皮或透黏膜贴剂、霜剂、软膏剂或乳剂。
所述药物组合物可以是无菌可注射水性或非水性(例如油质性)溶液剂或混悬剂的形式。所述无菌可注射制剂还可以是在无毒的肠胃外可接受的稀释剂或溶剂中的无菌可注射溶液剂或混悬剂,例如如1,3-丁二醇中的溶液剂。在可接受的载剂和溶剂中可使用的是水、磷酸盐缓冲溶液、林格溶液(ringer′ssolution)和等张氯化钠溶液。此外,按照惯例使用无菌的不挥发性油作为溶剂或助悬介质。出于此目的,可使用任何温和的不挥发性油,包括合成的甘油单酯或甘油二酯。此外,脂肪酸(例如油酸)可用于制备注射剂。混悬剂可根据已知技术使用那些合适的分散剂或润湿剂以及助悬剂来配制。
水性混悬剂包含活性成分,或在组合制剂的情况下包含多种活性成分,所述活性成分与适合于制备水性混悬剂的赋形剂混合。这样的赋形剂是助悬剂,例如羧甲基纤维素钠、甲基纤维素、羟丙基甲基纤维素、藻酸钠、聚乙烯吡咯烷酮、西黄蓍胶和阿拉伯树胶;分散剂或润湿剂,例如天然存在的磷脂(例如卵磷脂)、或环氧烷与脂肪酸的缩合产物(例如聚氧乙烯硬脂酸酯)、或环氧乙烷与长链脂族醇的缩合产物(例如十七乙烯氧基鲸蜡醇)、或环氧乙烷与来源于脂肪酸和己糖醇的偏酯(例如聚氧化乙烯与来源于脂肪酸和己糖醇酐的偏酯)的缩合产物(例如聚氧乙烯失水山梨糖醇单油酸酯)。所述水性混悬剂还可包含一种或更多种防腐剂(例如对羟基苯甲酸乙酯或对羟基苯甲酸正丙酯)、一种或更多种着色剂、一种或更多种矫味剂、以及一种或更多种甜味剂,例如蔗糖或糖精。
非水性(即油性)混悬剂可通过使活性成分混悬在植物油(例如花生油、橄榄油、芝麻油或椰子油)中或矿物油(例如液体石蜡)中来配制。所述油性混悬剂可包含增稠剂,例如蜂蜡、硬石蜡或鲸蜡醇。这些组合物可通过添加抗氧化剂(例如抗坏血酸)来保存。
适于通过添加水来制备水性混悬剂的可分散的散剂和颗粒剂提供了与分散剂或润湿剂、助悬剂和一种或更多种防腐剂混合的活性成分。合适的分散剂或润湿剂和助悬剂是已知的。
活性剂还可以以用于经直肠施用药物的栓剂的形式施用。这些组合物可通过使药物与合适的无刺激性赋形剂混合来制备,所述合适的无刺激性赋形剂在常温下是固体,但在直肠温度下是液体并将因此在直肠中融化以释放药物。这样的材料是可可豆脂和聚乙二醇。
对于局部递送,可使用经皮和透黏膜贴剂、霜剂、软膏剂、胶冻剂、溶液剂或混悬剂。对于舌下递送,可使用快速溶解的片剂制剂以及多种上述展示。对于经口施用,药物可作为片剂、胶囊剂或液体来施用。
制剂可方便地以单位剂量形式(例如,片剂和持续释放的胶囊剂)和脂质体存在,并且可通过药学领域中已知的任何方法来制备。药物制剂通常通过使活性物质或其可药用盐与常规的可药用载体、稀释剂或赋形剂混合来制备。赋形剂的一些实例是水、明胶、阿拉伯树胶、乳糖、微晶纤维素、淀粉、羟基乙酸淀粉钠、磷酸氢钙、硬脂酸镁、滑石、胶体二氧化硅等。这样的制剂还可包含其他药理学活性剂和常规添加剂,例如稳定剂、润湿剂、乳化剂、矫味剂、缓冲液等。通常来说,活性化合物的量为制剂的按重量计0.1至95%,优选地为肠胃外用制剂的按重量计0.2至20%且更优选地为经口施用制剂的按重量计1至50%。所述制剂还可通过已知方法(例如制粒、压制、微胶囊化、喷涂等)来制备。所述制剂可以过常规方法以片剂、胶囊剂、颗粒剂、散剂、糖浆剂、混悬剂、栓剂或注射剂的剂量形式来制备。液体制剂可通过使活性物质溶解或混悬在水或其他适合的载剂中来制备。片剂和颗粒剂可以以常规方式包被。为了长时间维持治疗有效的血浆浓度,可将本发明的化合物并入到缓慢释放制剂中。
特定化合物的剂量水平和剂量频率将根据多种因素而改变,这些因素包括所使用的特定化合物的效力、该化合物的代谢稳定性和作用时长、患者的年龄、体重、一般健康状况、性别、饮食、施用的模式和时间、排泄率、药物组合、待治疗病症的严重程度,以及正进行治疗的患者。每日剂量可例如为约0.001mg至约100mg/千克体重,以例如每次约0.01mg至约25mg的剂量单次或多次施用。这样的剂量可经口或肠胃外地给予。多剂量可在一段时间(例如至少一周、一个月、数月、一年或数年)中、或在整个病症过程中施用。剂量频率可以是至少每月一次、每周一次、或每天一次。
组合制剂
根据本发明的组合制剂的组分可用于同时、分开或依次使用。
本文中使用的术语“组合制剂”是指“套件(kitofparts)”,从这个意义上,(a)(r)-卡比多巴和(b)类固醇的组合组分可独立地或通过使用具有可分辨量的组合组分(a)和(b)的不同的固定组合来给药。所述组分可同时或相继地施用。如果所述组分相继地施用,优选地选择施用间的时间间隔以使得组合使用组分中对所治疗的障碍或疾病的作用大于可通过使用仅组合组分(a)和(b)中任一种所获得的作用。
组合制剂的组分可以以一种组合的单位剂量形式,或者作为组分(a)的第一单位剂量形式和组分(b)的单独的第二单位形式存在。可改变在组合制剂中待施用的组合组分(a)总量与组合组分(b)的比例,例如以便处理可由于例如患者的具体的疾病、年龄、性别或体重的待治疗的患者亚群的需要或单个患者的需要。
优选地,存在至少一种有益效果,例如增强(r)-卡比多巴抑制剂的效果,或相互增强组合组分(a)和(b)的效果,例如高于相加效果、另外的有利效果、更少的副作用、更低毒性、或与无效剂量的组合组分(a)和(b)之一或二者相比的组合治疗效果,以及非常优选地组合组分(a)和(b)的协同作用。
可向对象依次施用(r)-卡比多巴和类固醇,即可在类固醇之前、同时或之后施用(r)-卡比多巴。
(r)-卡比多巴和类固醇可在彼此的96小时、72小时、48小时、24小时或12小时之内施用。
或者,(r)-卡比多巴和类固醇可例如作为包含vap-1抑制剂和类固醇的组合物,或者通过同时施用单独剂量的(r)-卡比多巴和类固醇向对象共施用。
根据一些实施方案,向对象施用多个剂量的(r)-卡比多巴,和/或多个剂量的类固醇。
根据一些实施方案,在每次施用两个或更多个剂量的类固醇之前、同时或之后,施用一定剂量的(r)-卡比多巴。
例如,可在每次施用两个或更多个剂量的类固醇的96小时、72小时、48小时、24小时或12小时之内施用一定剂量的(r)-卡比多巴。
技术人员可例如通过观察患者(包括患者的整体健康状况和对组合治疗的响应)来确定和优化根据本发明的组合疗法中使用的组分的合适剂量的选择。例如,如果确定患者未表现出期望的治疗效果,或者相反地如果该患者经历数量过多或具有麻烦的严重程度的不期望或不良的副作用,则优化可以是必要的。
应该选择用于根据本发明的组合治疗的组分的剂量以在组合中提供治疗有效量的组分。组合治疗的“有效量”是导致与疼痛相关的至少一种病理学参数降低的量。例如,在一些实施方案中,组合治疗的有效量是与没有组合治疗时与疼痛相关的参数的期望降低相比,有效实现参数降低至少约10%、20%、30%、40%、50%、60%、70%、80%或90%的量。例如,参数可以是由根据西安大略省和麦克马斯特大学关节炎指数(westernontarioandmcmasteruniversitiesarthritisindex,womac)例如对于在行走、使用楼梯、在床上、坐或躺、以及站立期间的疼痛或日常活动、物理功能或僵硬评分的评估产生的评分。或者,参数可以是来自视觉模拟量表(visualanaloguescale,vas)、疼痛强度(painintensity,pi)量表、wong-baker面部疼痛评分量表、0-10数字疼痛评定量表、口头疼痛强度量表或描述符差异量表(descriptordifferentialscale)的评估的评分。
根据本发明,与(r)-卡比多巴或类固醇作为单一治疗的效果相比,可使用组合治疗以提高(r)-卡比多巴或类固醇的治疗效果,或降低所得的组合中单独组分的剂量,同时预防或进一步降低单独组分的不期望或有害副作用的风险。
对于在人中作为单一治疗的类固醇(特别是糖皮质激素,例如泼尼松或泼尼松龙),典型处方剂量范围是0.3至1mg/kg/天(合适地0.7或0.75mg/kg/天)或0.3mg/kg/天至10mg/kg/周。
对于在人中作为单一治疗的(r)-卡比多巴,典型处方剂量范围是20至200mg/天,通常30mg/天或75mg/天。
在一个实施方案中,(r)-卡比多巴和类固醇各自在对于作为单一治疗的每种化合物的典型处方剂量范围内的剂量下开处方。所述化合物可作为单独剂量或作为组合剂量开处方。与两者中任一种化合物作为单一治疗的效果相比,这样的组合提供了提高的效力。
在另一个实施方案中,(r)-卡比多巴和类固醇各自在低于对于作为单一治疗的每种组分的典型处方剂量的剂量下,但是具有组合治疗效力的剂量下开处方。所述组分可作为单独剂量或作为组合剂量开处方。可选择组合中组分的剂量以提供与(r)-卡比多巴或类固醇作为单一治疗相似水平的治疗效力,但与每种化合物作为单一治疗的处方剂量相比,具有(r)-卡比多巴和/或类固醇的更低剂量降低不良副作用风险的优点。
在另一个实施方案中,(r)-卡比多巴的处方剂量在对于单一治疗的典型处方剂量范围内,并且类固醇在低于对于单一治疗的典型处方剂量的剂量下开处方。
在另一个实施方案中,(r)-卡比多巴的处方剂量低于对于单一治疗的典型处方剂量,并且类固醇在对于单一治疗的典型处方剂量范围内的剂量下开处方。
低于对于单一治疗的典型处方剂量的优选剂量是典型处方剂量的高至50%或高至25%的剂量。
当以单独剂量施用时,(r)-卡比多巴和类固醇可基本上同时(例如,在彼此的约60分钟、约50分钟、约40分钟、约30分钟、约20分钟、约10分钟、约5分钟或约1分钟内)施用或在时间上隔开约1小时、约2小时、约4小时、约6小时、约10小时、约12小时、约24小时、约36小时、约72小时或约96小时或更多。
技术人员将能够根据(r)-卡比多巴和类固醇的特定组合来确定和优化依次施用的合适时间过程。优选地对时间过程进行选择,使得存在至少一种有益效果,例如增强(r)-卡比多巴或类固醇的效果,或相互增强组合组分的效果,例如高于相加效果、另外的有利效果、更少的副作用、更低毒性、或与无效剂量的组合组分之一或二者相比的组合治疗效果,以及非常优选地组合组分的协同作用。
应理解,最佳时间过程将取决于例如以下的因素:施用之后达到化合物峰血浆浓度所需的时间,以及每种化合物的消除半衰期。优选地时间差小于首个待施用组分的半衰期。
技术人员还将能够确定用于施用的合适时间。在特定的实施方案中,(r)-卡比多巴可以早晨施用,并且在一天中晚些时候施用类固醇至少一次。在另一些实施方案中,(r)-卡比多巴和类固醇可在基本上同一时间施用。
对象可在数周、数月或数年的时期内接受(r)-卡比多巴抑制剂和类固醇的剂量。例如,1周、2周、3周、1个月、2个月、3个月、4个月、5个月、6个月、7个月、8个月、9个月、10个月、11个月、1年、2年、3年、4年、5年、或更久。
通常来说,可通过已知的方式,在任何合适的制剂中,通过任何合适的途径来施用本发明组合的组分。合适的施用途径可包括经口、经直肠、经鼻、局部(包括口含和舌下)、舌下、经皮、鞘内、经黏膜或肠胃外(包括皮下、肌内、静脉内和皮内)施用。在一些实施方案中,经口施用(r)-卡比多巴抑制剂和类固醇。
合适的药物组合物和剂量形式可使用为药物制剂领域技术人员已知并且在相关的文本和文献(例如remington:thescienceandpracticeofpharmacy(easton,pa.:mackpublishingco.,1995))中描述的常规方法来制备。
以便于施用且剂量均一的单位剂量形式来配制本发明的组合制剂是尤其有利的。本文中使用的术语“单位剂量形式”是指适合作为用于待治疗个体的单一剂量的物理学上的离散单元。即,将组合物配制成离散剂量单元,其各自包含经计算以产生与所需药物载体相关的期望治疗效果的预定的“单位剂量”量的活性剂。本发明的单位剂量形式的规格取决于待递送的活性剂的独特特征。剂量还可通过参照成分的常用剂量和施用方式来确定。应注意到,在一些情况下,组合中两个或更多个单独的剂量单元提供了治疗有效量的活性剂,例如,两个片剂或胶囊剂一起可提供治疗有效剂量,使得在每个片剂或胶囊剂中的单位剂量为治疗有效量的约50%。
根据本发明用于肠胃外施用的制剂包括无菌的水性和非水性溶液剂、混悬剂和乳剂。可注射水性溶液剂包含水溶性形式的活性剂。非水性溶剂或载剂的实例包括脂肪油(例如橄榄油和玉米油)、合成脂肪酸酯(例如油酸乙酯或甘油三酯)、低分子量醇(例如丙二醇)、合成亲水性聚合物(例如聚乙二醇、脂质体)等。肠胃外制剂还可包含辅料(例如增溶剂、防腐剂、润湿剂、乳化剂、崩解剂和稳定剂),并且水性混悬剂可包含提高混悬剂黏度的物质,例如羧甲基纤维素钠、山梨糖醇和右旋糖酐。可注射制剂可通过引入灭菌剂、通过细菌保留过滤器过滤、辐照或加热而变得无菌。其还可使用无菌可注射介质来制备。活性剂还可以是干燥的(例如,冻干的)形式,其可在通过注射临施用之前用合适的载剂再水化。
除了先前描述的制剂之外,活性剂可配制成储库制剂(depotpreparation)用于活性剂的控制释放,优选地在长时期内持续释放。这些持续释放剂量形式通常通过植入(例如,皮下或肌内或者通过肌内注射)来施用。
本发明的组合制剂可与用于施用组合中组分的说明书一起包装。该说明书可记录在合适的记录介质或基质上,例如,该说明书可印刷在基质(例如纸或塑料)上。该说明书可作为包装插页(packageinsert)存在于容器或其组件的标签中(即,与包装或分包装缔合)。在另一些实施方案中,该说明书作为电子存储数据文件存在于合适的计算机可读存储介质(例如,cd-rom、磁盘)上。所述组合制剂的一些或所有组分可包装在合适的包装中以维持无菌性。
生物学数据
实施例1
人vap-1(ssao)抑制的体外确定
本测定在室温下用经纯化的重组表达的人vap-1(ssao)进行。基本上如ohman等(proteinexpressionandpurification46(2006)321-331)所述来制备酶。利用苄胺作为底物通过利用辣根过氧化物酶(horseradishperoxidise,hrp)偶联反应中过氧化氢的产生测定酶活性。简单地说,使受试化合物溶解在二甲基亚砜(dimethylsulfoxide,dmso)中至浓度为10mm。通过在dmso中产生1∶10连续稀释以产生7点曲线或通过在dmso中制备1∶3连续稀释以产生11点曲线来测定剂量响应测量。根据化合物的效力调整最高浓度并且随后在反应缓冲液中的稀释产生≤2%的最终dmso浓度。
在辣根过氧化物酶(hrp)偶联反应中,10-乙酰基-3,7-二羟基吩
使用本测定,通过(s)-卡比多巴和(r)-卡比多巴对人vap-1的抑制的ic50分别为142nm和148nm。
实施例2
人芳香族l-氨基酸脱羧酶(多巴脱羧酶)抑制的体外确定
根据制造说明书(r&d系统cat#3564-dc)进行人多巴脱羧酶的酶反应。该测定测量了重组人多巴脱羧酶使l-多巴转换成多巴胺的能力。用三硝基苯硫酸衍生化之后,通过其在340nm处的吸光度测量多巴胺产物。简单地说,在含有100mmnacl的50mmhepes缓冲液(ph7.2)中进行反应30分钟,并通过在95℃下2分钟使酶失活而中止反应。通过用0.8μg酶、500μml-多巴和100μm吡哆醛磷酸盐,在存在或不存在固定量的每种化合物的情况下测量多巴脱羧酶的脱羧酶活性来进行抑制测定。受试化合物与多巴脱羧酶一起预孵育60分钟,随后通过添加底物l-多巴开始该测定。生成剂量反应曲线以确定抑制50%脱羧酶活性所需的浓度(ic50)。化合物以7种浓度重复评价。通过非线性回归分析得到ic50值。
使用本测定,通过(s)-卡比多巴对人芳香族l-氨基酸脱羧酶的抑制的ic50为190nm,然而直至最大测试浓度(3μm),(r)-卡比多巴并不抑制人芳香族l-氨基酸脱羧酶。
实施例3
在大鼠中单独的和与泼尼松龙组合的(r)-卡比多巴二者对cfa(completefreundsadjuvant,完全弗氏佐剂)诱导的超敏反应的作用
(r)-卡比多巴的抗痛觉过敏特性的评估通过测量cfa诱导超敏反应之后的负重来确定。未经处理大鼠(naiverat)在两只后爪间同等地分布其体重。然而,当经注射的(左)后爪疼痛时,体重重新分布使得更轻的重量放在受影响的爪上(在受伤爪上的负重降低)。使用大鼠失能测量仪(ratincapacitancetester)(lintoninstruments,uk)测量通过每一条后肢的负重。将大鼠放置在失能测量仪中,后爪放在单独的传感器上并且在4秒中记录两条后肢所施加的平均力。cfa的注射还诱导水肿,其可通过爪体积进行评估;这使用容积测量仪(plethysmometer)进行测量。将大鼠的后爪放置在含有溶液的圆柱体中,并且被置换的液体体积确定了爪体积。
用随意可获得的食物和水使未经处理雄性spraguedawley大鼠适应。进行对失能测量仪的习惯化。在诱导损伤之前进行基线负重和爪体积记录。通过将cfa(100μl的1mg/ml溶液)足底内注射到左后爪中来诱导炎性超敏反应。cfa之后23小时进行处理前负重和爪体积测量以评估超敏反应。然后根据拉丁方设计(latinsquaredesign)中的cfa窗对动物进行排序和随机化。
在a部分,在cfa之后24小时,用载剂(5%dmso、0.5%羟丙基甲基纤维素(hpmc)于水中),(r)-卡比多巴3mg/kg、10mg/kg和30mg/kg,或吲哚美辛10mg/kg(10ml/kg剂量体积)处理动物。在处理之后1小时和3小时测量负重,并且在处理之后3小时测量水肿。
在b部分:在cfa之后24小时,用载剂(1%甲基纤维素(mc)于水中),泼尼松龙0.3mg/kg、1mg/kg、3mg/kg和10mg/kg,或吲哚美辛10mg/kg(5ml/kg剂量体积)处理动物。在处理之后1小时和3小时测量负重。
在c部分,在cfa之后24小时处理动物两次,用载剂(5%dmso0.5%hpmc)或(r)-卡比多巴3mg/kg和10mg/kgp.o.(10ml/kg剂量体积)进行一次,并随后用载剂(1%mc)或泼尼松龙0.3mg/kgp.o.(5ml/kg剂量体积)。在处理之后1小时和3小时测量负重,并且在处理之后3小时测量水肿。
通过在每个时间点比较处理组和载剂对照组分析数据。
对右后爪和左后爪二者进行负重(g)读数并计算差异。数据表示为超敏反应至疼痛的逆转%。对左后爪进行爪体积(ml)读数。数据表示为水肿的逆转%。计算:(给药之后的读数-给药之前的读数)/(未经处理的读数-给药之前的读数)×100,其中未经处理的负重差异-给药之前负重差异被定义为待逆转的cfa窗。通过重复测量anova、随后使用invivostat(invivostat.co.uk)进行计划比较检验(plannedcomparisontest)来进行统计学分析,(p<0.05被认为是显著的)。
结果
如给药之后24小时受伤与未受伤后爪之间的负重转变所检测的,cfa的足底内注射诱导了超敏反应。在这两个研究中,cfa还在经注射的爪中诱导了显著的水肿。与先前的研究一致,吲哚美辛(10mg/kg)产生了使用负重测量的超敏反应的显著逆转。吲哚美辛还表现出爪水肿的显著降低。
a部分:单独的(r)-卡比多巴(3至30mg/kg)剂量依赖地抑制超敏反应应答(参见图1)并且显示了爪体积的显著降低(参见图2)。
b部分:单独的泼尼松龙(0.3至10mg/kg)剂量依赖地抑制超敏反应应答(参见图3)。
c部分:选择(r)-卡比多巴(3和10mg/kg)和泼尼松龙(0.3mg/kg)的最低/适度有效剂量进行组合施用以评价潜在的协同作用。
泼尼松龙(0.3mg/kg)和(r)-卡比多巴的共给药具有与单独的3mg/kg泼尼松龙相同的镇痛作用,表明当与(r)-卡比多巴共给药时,类固醇的给药可降低超过10倍(参见图4)。
结果还示出泼尼松龙与(r)-卡比多巴之间的协同作用的证据(参见图4)。
可根据参考文献[1]和[2]中所教导的方法计算协同作用:
[1]webbjl,effectofmorethanoneinhibitor.enzymeandmetabolicinhibitors.1.newyork:academicpress;1963,p.66-79(488-512)
[2]grecowr,bravog,andparsonsjc(1995)thesearchforsynergy:acriticalreviewfromaresponsesurfaceperspective.pharmacolrev47:331-385.
- 还没有人留言评论。精彩留言会获得点赞!
- 检测分析人体脓毒症的方法
- 一种胶乳颗粒增强型中性粒细胞明胶酶相关脂质运载蛋白检测试剂盒的制作方法
- 免疫激活剂的制作方法
- 具有取代苯基硫属原子代低级烷基和导入酯结构的苯基作为取代基的新型1,2-二氢喹啉 ...的制作方法
- 具有(取代苯基或取代杂环)羰基氧基低级烷基和导入了酯结构的苯基作为取代基的新型 ...的制作方法
- 具有糖皮质激素受体结合活性的新型1,2,3,4-四氢喹喔啉衍生物的制作方法
- 治疗心血管疾病,炎症和免疫病症的化合物及方法
- 具有糖皮质激素受体结合活性且具有导入了磺酸酯或磺酰胺结构的苯基作为取代基的新 ...的制作方法
- 具有IκB激酶β抑制活性的新型吲哚衍生物的制作方法
- Toll样受体9阻断剂及应用的制作方法