GnRH类似物-抗肿瘤药物偶联物、其制备方法及用途与流程
本发明涉及属于医药化学领域,具体涉及一种gnrh类似物-抗肿瘤药物偶联物,还涉及其制备方法与用途。
背景技术:
临床用于治疗肿瘤的化学药物由于缺乏选择性,在杀伤肿瘤细胞的同时也会杀伤正常组织细胞,引起严重的毒副作用,给患者造成极大的痛苦,加之易引发耐药性等缺点,化疗药物的临床应用受到很大限制。因此,寻找新型高效低毒的抗癌药物成为肿瘤创新药物治疗研究中极具挑战的难题之一。
人促性腺激素释放激素(gonadotropin-releasinghormone,gnrh)是下丘脑分泌的一种多肽类激素,其氨基酸序列为pyr1-his2-trp3-ser4-tyr5-gly6-leu7-arg8-pro9-gly10-nh2(pehwsyglrpg-nh2),与脑垂体上的gnrh受体特异性结合后,刺激垂体产生黄体生成素(lh)和卵泡刺激素(fsh),进而调节性激素的分泌及生殖功能。研究表明,gnrh受体不仅在脑垂体表达,其在乳腺癌、卵巢癌、前列腺癌等肿瘤细胞表面均呈现过表达状态,而在正常组织的表达量很低,这使得gnrh受体作为抗肿瘤药物靶向给药的作用靶点成为可能。
多肽-药物偶联物(peptide-drugconjugates,pdc)是将抗肿瘤药物与肿瘤细胞靶向多肽载体通过适当的连接臂偶联在一起得到的复合物,是目前肿瘤药物学研究方向的热点之一,有可能成为肿瘤治疗的新途径。目前,在以gnrh受体为靶标的多肽-药物偶联物研究中,应用较多的gnrh受体靶向肽主要是gnrh类似物[d-lys6]-gnrh或[gln1,d-lys6,des-gly10,pro9-nhet]-gnrh,将其与抗肿瘤药物通过酰胺键、酯键等连接臂偶联,获得了一些活性较好的靶肽-药物偶联物。an-152是一个将[d-lys6]-gnrh与阿霉素通过戊二酸酐连接的多肽-药物偶联物,因其良好的靶向抗肿瘤活性已经进入临床研究阶段,然而an-152的血清半衰期为2.1小时,用药过程中会产生一定的毒副作用。
因此,仍需要研究新的gnrh类似物-抗肿瘤药物偶联物,在保持较高的肿瘤细胞增殖抑制活性的同时,提高偶联物的稳定性,对于降低药物的毒副作用具有重要的意义。
技术实现要素:
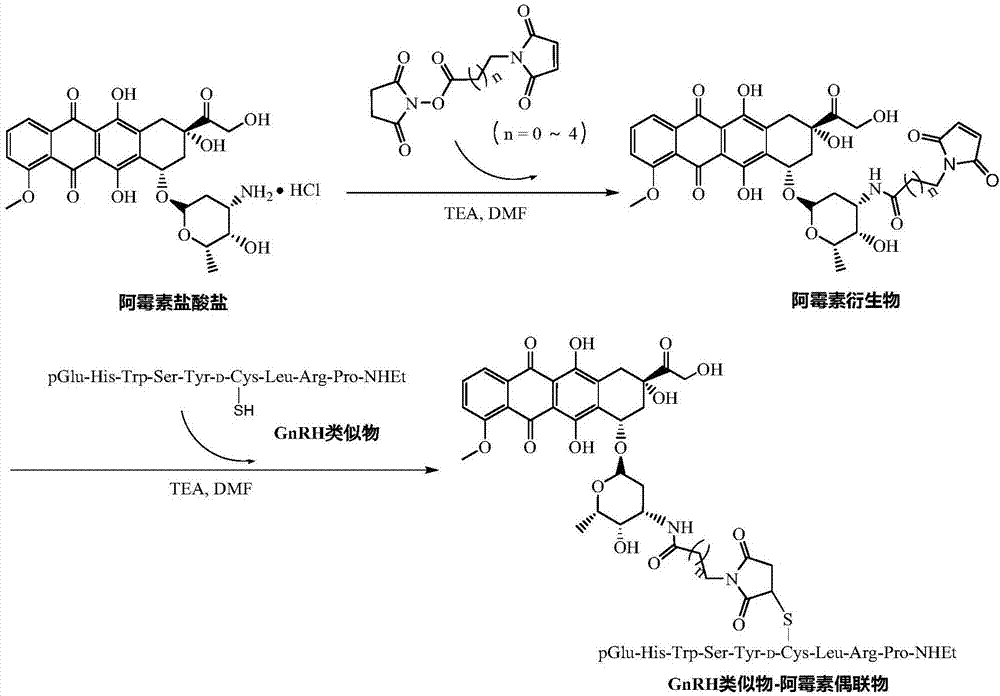
为了解决现有gnrh类似物-抗肿瘤药物偶联物稳定性不高,容易过早的释放出细胞毒分子,进而产生毒副作用等问题,本发明提出一种gnrh的结构修饰策略,即将gnrh中的gly6用d-cys取代,同时c末端pro9乙胺酰胺化,得到一种新的gnrh类似物,表示为[d-cys6-desgly10-pro9-nhet]-gnrh。将抗肿瘤药物阿霉素通过马来酰亚胺基和羟基琥珀酰亚胺酯双功能基团的连接臂与上述gnrh类似物偶联,制备具有较高的肿瘤细胞增殖抑制活性、较强的gnrh受体靶向能力,同时稳定性得到提高的gnrh类似物-药物偶联物。
为了实现上述目的,本发明提供一种gnrh类似物-抗肿瘤药物偶联物,所述偶联物具有如下所示的结构:
gnrha-linker-dox
其中,gnrha为gnrh类似物,其肽序列为pglu1-his2-trp3-ser4-tyr5-d-cys6-leu7-arg8-pro9-nhet;linker为连接臂,用于将gnrha与抗肿瘤药物偶联;dox为抗肿瘤药物阿霉素。
在本发明中,所述连接臂具有下式所示的结构:
其中n=0、1、2、3、4,优选地,n=1。
因此,所述偶联物具有下式所示的结构:
其中n=0、1、2、3、4。
本发明还提供上述gnrh类似物-抗肿瘤药物偶联物的制备方法,所述方法包括以下步骤:
(1)制备[d-cys6-desgly10-pro9-nhet]-gnrh:
采用固相多肽合成法,在n,n'-二异丙基乙胺(dipea)存在的条件下由9-芴甲氧羰基保护氨基的脯氨酸(fmoc-pro-oh)和取代度为0.6~1.3mmol/g的2-ctc树脂反应得到fmoc-pro-ctc树脂,其中2-ctc树脂与dipea和fmoc-pro-oh的摩尔比为1:2~6:2~6;所得fmoc-pro-ctc树脂经过体积浓度为20%~40%的哌啶/n,n'-二甲基甲酰胺(dmf)溶液除去fmoc保护基团后,以n,n'-二异丙基碳二亚胺(dic)/1-羟基苯并三唑(hobt)为缩合试剂,按照肽序列从c端向n端逐步偶联氨基酸得到全保护肽树脂,其中fmoc-pro-ctc树脂、缩合试剂dic/hobt和fmoc-氨基酸的摩尔比为1:2~6/2~6:2~6,偶联时间为1~3h;按照质量体积比为1g:5~10ml的比例向所述全保护肽树脂中添加体积浓度为0.5~3.5%三氟乙酸(tfa)的二氯甲烷(dcm)中溶液切肽;所得全保护肽用质量体积比为1g:5~10ml的dcm溶解,通过dipea调节体系ph为8~10,加入六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷(pybop)和乙胺盐酸盐,其中全保护肽与pybop和乙胺盐酸盐的摩尔比为1:1~3:1~3,hplc监控反应进程,反应完毕后将体系减压浓缩,按照质量体积比为1g:5~10ml的比例向所得产物中加入体积比为90/5/3/2的tfa/甲基苯基硫醚/1,2-乙二硫醇(edt)/苯甲醚的裂解液脱除侧链保护基团,室温反应1~3h后将体系减压浓缩,然后用8~10倍于反应浓缩液体积的冰乙醚沉降、洗涤,所得固体用反相高效液相色谱纯化、冷冻干燥后得到[d-cys6-desgly10-pro9-nhet]-gnrh纯品;
(2)制备阿霉素衍生物
将阿霉素盐酸盐(dox·hcl)、异双功能基团连接臂和三乙胺(tea)用无水dmf溶解,其中dox·hcl、异双功能基团连接臂和tea的摩尔比为1:1.0~2.0:2.0~3.0,室温搅拌反应1~3h;反应完毕后用2~5倍于反应液体积的冰乙醚沉降,所得沉淀用冰乙醚洗涤,真空恒温干燥得阿霉素衍生物;
(3)[d-cys6-desgly10-pro9-nhet]-gnrh与阿霉素衍生物的偶联
将步骤(1)得到的[d-cys6-desgly10-pro9-nhet]-gnrh与步骤(2)得到的阿霉素衍生物用dmf溶解,以tea为催化剂,其中[d-cys6-desgly10-pro9-nhet]-gnrh、阿霉素衍生物和tea的摩尔比为1:1.0~2.0:15.0~25.0;n2保护下室温搅拌反应1~3h,反应完毕后用8~10倍于反应液体积的冰乙醚沉降、洗涤,所得固体用反相高效液相色谱纯化、冷冻干燥后得到所述gnrh类似物-抗肿瘤药物偶联物。
在本发明中,所述的异双功能基团连接臂选自2-马来酰亚胺基乙酸羟基琥珀酰亚胺酯、3-马来酰亚胺基丙酸羟基琥珀酰亚胺酯、4-马来酰亚胺基丁酸羟基琥珀酰亚胺酯、5-马来酰亚胺基戊酸羟基琥珀酰亚胺酯或6-马来酰亚胺基己酸羟基琥珀酰亚胺酯。
本发明还涉及含有上述偶联物的药物组合物。
本发明还提供上述gnrh类似物-抗肿瘤药物偶联物在制备用于预防或治疗肿瘤的药物中的用途。其中,所述肿瘤是乳腺癌、前列腺癌、卵巢癌、宫颈癌、子宫内膜癌。
实验证实,本发明的gnrh类似物-抗肿瘤药物偶联物在血清中的稳定性有所增强;另外,该偶联物对gnrh受体过表达的肿瘤细胞(如人乳腺癌细胞)具有较强的增殖抑制活性,而且这种增殖抑制活性是由gnrh受体介导的内吞作用实现的;同时该偶联物对gnrh受体表达阴性的细胞(如3t3小鼠胚胎成纤维细胞)毒性较小,因此具有良好的肿瘤细胞选择性。上述结果表明本发明所采用的gnrh结构改造策略是切实可行的,本发明第一方面所述的偶联物有利于降低阿霉素的毒副作用,为新的gnrh受体靶向抗肿瘤药物的研发奠定了基础,具有良好的应用前景。
【附图说明】
图1gnrha-linker-dox偶联物的合成路线图
图2dox-smp的1h-nmr谱图
图3gnrha-linker-dox和dox对mcf-7人乳腺癌细胞的增殖抑制活性
图4gnrha-linker-dox和dox的gnrh受体抑制实验结果
图5gnrha-linker-dox和dox对3t3小鼠胚胎成纤维细胞的毒性
【具体实施方式】
在本发明中使用的缩写具有下面的含义:
gnrh促性腺激素释放激素
dox·hcl阿霉素盐酸盐
smp3-马来酰亚胺基丙酸羟基琥珀酰亚胺酯
fmoc9-芴甲氧羰基
pro脯氨酸
arg精氨酸
leu亮氨酸
cys半胱氨酸
tyr酪氨酸
ser丝氨酸
trp色氨酸
his组氨酸
pglu焦谷氨酸
pbf2,2,4,6,7-五甲基二氢苯并呋喃-5-磺酰
trt三苯甲基
tbu叔丁基
boc叔丁氧羰基
dicn,n'-二异丙基碳二亚胺
hobt1-羟基苯并三唑
dipean,n'-二异丙基乙胺
pybop六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷
tfa三氟乙酸
tea三乙胺
edt1,2-乙二硫醇
dmfn,n'-二甲基甲酰胺
dmso二甲基亚砜
dcm二氯甲烷
hplc高效液相色谱
esi-ms电喷雾质谱
1h-nmr核磁共振氢谱
以下实施例用于非限制性地解释本发明的技术方案。
在以下实施例中,2-ctc树脂购自天津南开合成科技有限公司;fmoc-pro-oh、fmoc-arg(pbf)-oh、fmoc-leu-oh、fmoc-d-cys(trt)-oh、fmoc-tyr(tbu)-oh、fmoc-ser(tbu)-oh、fmoc-trp(boc)-oh、fmoc-his(trt)-oh和pyroglutamicacid为成都诚诺新科技有限公司产品;dic、hobt、dipea和pybop购自苏州昊帆生物科技有限公司;dox·hcl、乙胺盐酸盐为上海阿拉丁生化科技股份有限公司产品;smp购自梯希爱化成工业发展有限公司;tfa、甲基苯基硫醚(thioanisole)和苯甲醚(anisole)购自百灵威科技有限公司;edt为amresco公司产品;dmf、dcm、哌啶、tea和乙醚等均为市售分析纯试剂;甲醇、乙腈为市售色谱纯试剂。
实施例1[d-cys6-desgly10-pro9-nh2]-gnrh的合成:
2-ctc树脂(取代度1.3mmol/g,5mmol)与fmoc-pro-oh(15mmol)在dipea作用下反应得到fmoc-pro-2-ctc树脂,其中2-ctc树脂与dipea和fmoc-pro-oh的摩尔比为1:3:3;所得fmoc-pro-2-ctc树脂用体积浓度为20%的哌啶/dmf溶液脱除fmoc保护基团,茚三酮检测呈阳性,表明fmoc保护基团被成功脱除;应用fmoc/tbu交叉保护策略,以dic(22.5mmol)/hobt(22.5mmol)为缩合试剂,按照标准merrifield多肽固相合成法从c端向n端依次缩合剩余fmoc-氨基酸(22.5mmol),其中fmoc-pro-ctc树脂、缩合试剂dic/hobt和fmoc-氨基酸的摩尔比为1:4.5/4.5:4.5;反应时间2h,得到[d-cys6-desgly10-pro9-oh]-gnrh全保护肽树脂。按照质量体积比为1g:10ml的比例向所述全保护肽树脂中添加体积浓度为2%的tfa/dcm溶液切肽;所得全保护肽用质量体积比为1g:5ml的dcm溶解,dipea调节体系ph为9,加入1.5倍摩尔量的pybop和1.5倍摩尔量的乙胺盐酸盐,hplc监控反应进程,反应2h后将体系减压浓缩,按照质量体积比为1g:10ml的比例向所得产物中加入配比为tfa/甲基苯基硫醚/edt/苯基醚(90/5/3/2,v/v)的裂解液脱除侧链保护基团,室温反应2h后将反应液浓缩,然后用10倍于反应浓缩液体积的冰乙醚沉降、洗涤三次,所得固体经真空干燥至恒重后用反相高效液相色谱纯化、冷冻干燥后得到[d-cys6-desgly10-pro9-nh2]-gnrh纯品,hplc分析纯度为98%;esi-ms:m/z,[m+h]+:1200.4(理论值),1199.8(实验值);[m+2h]2+:600.7(理论值),600.8(实验值)。
实施例2
阿霉素与3-马来酰亚胺基丙酸羟基琥珀酰亚胺酯(smp)的反应
参照文献shinq,gaow,xiangb,etal.enhancingcellularuptakeofactivablecell-penetratingpeptide-doxorubicinconjugatebyenzymaticcleavage[j].intjnanomedicine.2012,7:1613–1621的记载,称取dox·hcl(227mg,0.39mmol)与smp(114.5mg,0.43mmol),用无水dmf(40ml)溶解,再加入tea(125μl,0.90mmol),室温搅拌反应2h后将反应液用冰乙醚(100ml)沉降,离心后沉淀用冰乙醚洗涤(50ml×3),真空恒温干燥得174.5mg阿霉素衍生物(dox-smp,产率64%),用hplc、esi-ms和1h-nmr(图2)予以表征。
hplc分析纯度为96%;esi-ms:m/z,[m+na]+:717.6(理论值),717.2(实验值);1h-nmr(300hz,dmso-d6):3.92ppm处的单峰代表4位-och3的3个氢原子,4.56ppm处的单峰代表14位-ch2-的2个氢原子,6.95ppm的单峰代表马来酰亚胺基上的1”-h和2”-h,7.6-7.9ppm处的几个单峰分别代表苯环上1-3位的3个氢原子,13.26ppm和14.02ppm处的单峰分别代表代表11位和6位-oh的氢原子,以上特征官能团的氢化学位移值表明了dox-smp的成功合成。
实施例3gnrha-linker-dox偶联物的合成:
实施例1得到的gnrha(33.6mg,28μmol)与实施例2得到的dox-smp(20mg,28μmol)用无水dmf(10ml)溶解,再加入tea(80μl,577μmol),其中gnrha与dox-smp和tea的摩尔比为1:1:21;n2保护下室温搅拌反应,hplc监控反应进程,反应完毕后将反应液用冰乙醚(50ml)沉降,离心后沉淀用冰乙醚洗涤(20ml×3),最后经半制备高效液相色谱纯化、冷冻干燥后得到目标偶联物gnrha-linker-dox。esi-ms:m/z,[m+h]+:1895.0(理论值),1894.9(实验值);[m+2h]2+:948.0(理论值),947.8(实验值)。
实施例4gnrha-linker-dox偶联物的血清稳定性:
dmso溶解实施例3得到的gnrha-linker-dox偶联物,加入人血清稀释,涡旋均匀,得到待测样品溶液(gnrha-linker-dox偶联物的终浓度为100μm,dmso体积比为0.1%),37℃恒温振荡器中孵育,不同时间点取样100μl,三倍体积乙腈终止反应;高速离心(10000×g)10min,取20μl上清液进行hplc分析,以0h孵育体系中偶联物的峰面积积分值为100%,根据不同取样时间点孵育体系中偶联物的峰面积积分值计算其原型保留百分含量。
按照上述方法,血清稳定性测定结果显示:所述偶联物在血清中表现出了较高的稳定性,半衰期为7.5h,明显高于现有的多肽-药物偶联物an-152。
实施例5肿瘤细胞增殖抑制活性实验
mcf-7人乳腺癌细胞培养于含10%胎牛血清、100u/ml青霉素、链霉素的dmem培养液中,在恒温37℃,5%co2培养箱中培养,隔3天传代,取对数生长期细胞接种于96孔板(约5000个细胞/孔)中,孵育24h。
采用mtt法测定dox与gnrha-linker-dox偶联物对mcf-7人乳腺癌细胞的体外增殖抑制活性。dmso分别溶解dox与gnrha-linker-dox偶联物,用无血清dmem培养基稀释得到梯度浓度药液(25,50,75,100μm),分别取不同浓度的药液100μl加入到各孔中,37℃孵育48h后,每孔加入10μl的mtt溶液(5mg/ml),于37℃孵育4h后弃去上清液,每孔加入150μldmso,在570nm处用酶标仪测量每个孔的od值,根据公式计算抑制率。
肿瘤细胞增殖抑制活性结果表明,gnrha-linker-dox偶联物对mcf-7人乳腺癌细胞表现出较高的增殖抑制活性(图3),且该增殖抑制活性具有浓度依赖性。
实施例6gnrh受体抑制实验
通过gnrh受体抑制实验考察gnrha-linker-dox偶联物的肿瘤细胞靶向性。以gnrh受体过表达的mcf-7细胞为受试对象,取对数生长期细胞并将其分成两组,接种于96孔板(约5000个细胞/孔)中,在37℃、5%co2恒温培养箱中孵育24h。其中一组加入含亮丙瑞林(10μm)的培养基溶液预处理,另一组加入空白培养基,孵育2h后吸除培养基,加入dox和gnrha-linker-dox偶联物的药物溶液,按照实施例5的mtt实验方法计算药物对mcf-7细胞的抑制率,分别考察gnrha-linker-dox偶联物和dox对两组mcf-7细胞的增殖抑制活性差异。
gnrh受体抑制实验结果显示:gnrha-linker-dox偶联物对亮丙瑞林预处理组mcf-7细胞(其gnrh受体被亮丙瑞林预先占据)的增殖抑制活性低于未经亮丙瑞林处理组细胞的增殖抑制活性(图4a,其中*p<0.05,**p<0.01),而阿霉素对mcf-7细胞的增殖抑制活性却不受亮丙瑞林作用的影响(图4b),表明该偶联物的肿瘤细胞增殖抑制活性是通过gnrh受体介导进入细胞而实现的,这为偶联物的细胞选择性奠定了基础。
实施例7gnrha-linker-dox偶联物的细胞毒性
以gnrh受体阴性表达的nih/3t3小鼠胚胎成纤维细胞为受试对象,考察gnrha-linker-dox偶联物的细胞毒性。nih/3t3细胞培养于含10%新生牛血清、100u/ml青霉素、链霉素的dmem培养液中,在恒温37℃、5%co2培养箱中培养,隔2~3天传代,取对数生长期细胞接种于96孔板(约5000个细胞/孔)中,孵育24h。按照实施例5中的mtt法测定dox与gnrha-linker-dox偶联物对3t3细胞的抑制率。
按照上述方法,dox与gnrha-linker-dox偶联物对3t3细胞的细胞毒活性结果见图5。由于dox缺乏选择性,其对3t3细胞具有较高的毒性杀伤作用;但是dox与gnrh类似物[d-cys6-des-gly10-pro9-nhet]-gnrh偶联后,得到的本发明的gnrha-linker-dox偶联物对3t3细胞的毒性显著降低,这一结果与3t3细胞不表达gnrh受体,偶联物不能通过gnrh受体介导进入细胞有关,进一步验证[d-cys6-des-gly10-pro9-nhet]-gnrh对gnrh受体的靶向结合能力,同时也说明本发明所采用的gnrh结构改造策略是切实可行的,为新型gnrh受体靶向的抗肿瘤药物研发奠定了基础,具有良好的应用前景。
- 还没有人留言评论。精彩留言会获得点赞!