帕洛诺司琼透皮贴剂及其制备方法与流程
本发明属于医药技术领域,特别的涉及一种帕洛诺司琼透皮贴剂及其制备方法。
背景技术:
癌症化疗在杀死癌细胞同时也造成正常细胞的毒性,从而引起患者在化疗后难以忍受的痛苦。化学疗法引起的恶心和呕吐(cinv)仍然是主要的副作用,尽管有大量新型止吐药,通常观察到延迟效应。
帕洛诺司琼是第二代高选择性、高亲和性的5-ht3受体拮抗剂,与第一代5-ht3受体拮抗剂相比有更长半衰期,被美国fda批准用于防治化疗引起的迟发性恶心呕吐。目前该药物的市售制剂主要为注射剂和胶囊剂,但注射剂给药不方便、顺应性差且多次注射易导致静脉炎,口服胶囊剂在治疗呕吐上不利于给药且不利于吞咽有困难的患者使用。透皮贴剂在给药方便,维持长效平稳的血药浓度及提高患者顺应性等方面展现良好优势。
帕洛诺司琼的分子量是296,logp为2.7,以及熔点是91℃,使其便于通过经皮给药的方式进入血液循环系统,符合开发成经皮给药系统的基本要求。
欧洲专利ep2286814b1公开了在帕洛诺司琼透皮贴剂中加入脂肪醇类添加剂以抑制其结晶;沈阳药科大学专利cn-104069505b采用帕洛诺司琼碱的β环糊精的包合物制成透皮贴剂其发明目的是七天给药,7天缓慢释放。
以上文献公开的帕洛诺司琼透皮贴剂均是从药物结晶角度设计开发的,本发明是在贴剂中加入聚丙烯酸树脂提高药物稳定性和经皮透过率,发明人惊奇的发现由于帕洛诺司琼在人体内的半衰期是40~44h,可以直接通过快速释放而不用包合来达到透过皮肤达到长效给药的目的,并且本发明是通过加入聚丙烯酸树脂提高药物的释放速率和经皮透过率,使其贴于皮肤1~3天,达到5~7天的给药需求。
技术实现要素:
本发明的目的在于提供一种制备简单,有良好稳定性、不用包合,1~3天给药而达到5~7天的给药需求和经皮渗透率的帕洛诺司琼透皮贴剂。
本发明的目的之二是提供一种帕洛诺司琼透皮贴剂的制备方法。
本发明的这些以及其它目的将通过下列详细描述和说明来进一步的体现。
本发明的帕洛诺司琼透皮贴剂,由背衬层、载药压敏胶层以及防粘层构成,所述的载药压敏胶层包括帕洛诺司琼碱、聚丙烯酸树脂、聚丙烯酸酯类压敏胶、化学促渗剂以及抗氧剂,其中帕洛诺司琼碱占载药压敏胶层总重的1%~5%,贴剂的含药量在0.5~20.0mg,最好在0.5~2.0mg,贴剂面积≤10cm2。
在本发明的帕洛诺司琼透皮贴剂中,载药压敏胶层包括按重量份计的以下组分:
进一步的,在本发明的帕洛诺司琼透皮贴剂中,载药压敏胶层包括按重量份计的以下组分:
较好的是,在本发明的帕洛诺司琼透皮贴剂中,载药压敏胶层包括按重量份计的以下组分:
在本发明的帕洛诺司琼透皮贴剂中,所述的聚丙烯酸树脂为e100和/或rlpo;所述的化学促渗剂为ipm,所述的抗氧剂为维生素e,所述的压敏胶为聚丙烯酸压敏胶。
在本发明的帕洛诺司琼透皮贴剂中,所述的聚丙烯酸树脂是甲基丙烯酸二甲氨基乙酯、甲基丙烯酸、甲基丙烯酸酯等单体按不同比例共聚产生。
本发明的帕洛诺司琼透皮贴剂的制备方法,包括如下步骤:
(1)将聚丙烯酸树脂用乙酸乙酯溶胀后加入聚丙烯酸酯压敏胶搅拌至均匀,形成压敏胶基质;
(2)向压敏胶基质中加入化学促渗剂和抗氧剂,搅拌均匀后加入帕洛诺司琼碱的药物溶液,在室温下搅拌;
(3)形成均一的载药压敏胶溶液后,放在室温下静置0.5~1.5小时脱气,将载药压敏胶基质涂布在防粘层上,烘干乙酸乙酯,复合上背衬层,切成合适尺寸,形成帕洛诺司琼透皮贴剂。
进一步的,本发明的帕洛诺司琼透皮贴剂,由背衬层、载药压敏胶层以及防粘层构成,其中帕洛诺司琼碱占载药压敏胶层总重的1%~5%,贴剂的含药量在0.5~20mg,较好的是0.5~2.0mg,贴剂面积<10cm2,所述的载药压敏胶层包括按重量份计的以下组分:
按以下方法制备:
(1)将聚丙烯酸树脂用乙酸乙酯溶胀后加入聚丙烯酸酯压敏胶搅拌至均匀,形成压敏胶基质;
(2)向压敏胶基质中加入化学促渗剂和抗氧剂,搅拌均匀后加入帕洛诺司琼碱的药物溶液,在室温下搅拌;
(3)形成均一的载药压敏胶溶液后,放在室温下静置一小时脱气,将载药压敏胶基质涂布在防粘层上,烘干乙酸乙酯,复合上背衬层,切成合适尺寸,形成帕洛诺司琼透皮贴剂。
在本发明中帕洛诺司琼碱可以采用以下方法制备:
称取一定质量的盐酸帕洛诺司琼,加水溶解,加过量0.5mol/l氢氧化钠溶液,使得帕洛诺司琼碱完全产生并沉淀,用抽滤的方式过滤沉淀,并用水洗至中性,在真空干燥箱中干燥后,装瓶保存。
也可以采用cn201410315219.0所述的方法或其他方法制备得到,但这些方法得到的帕洛诺司琼碱没有本发明的方法好。
实验发现当丙烯酸酯压敏胶中不含聚丙烯酸树脂时,单一未知杂质含量约为总重量的0.4%;加入总重量30%的聚丙烯酸树脂后,未知杂质含量降低到总重量的0.1%左右;并且在大鼠体外透过实验中,与未加聚丙烯酸树脂贴剂相比,加入不同比例聚丙烯酸树脂均能提高贴剂的经皮透过率。
另外,本发明的贴剂生产工艺简单,生产成本低,给药1~3天能维持5~7天的药效。
附图说明:
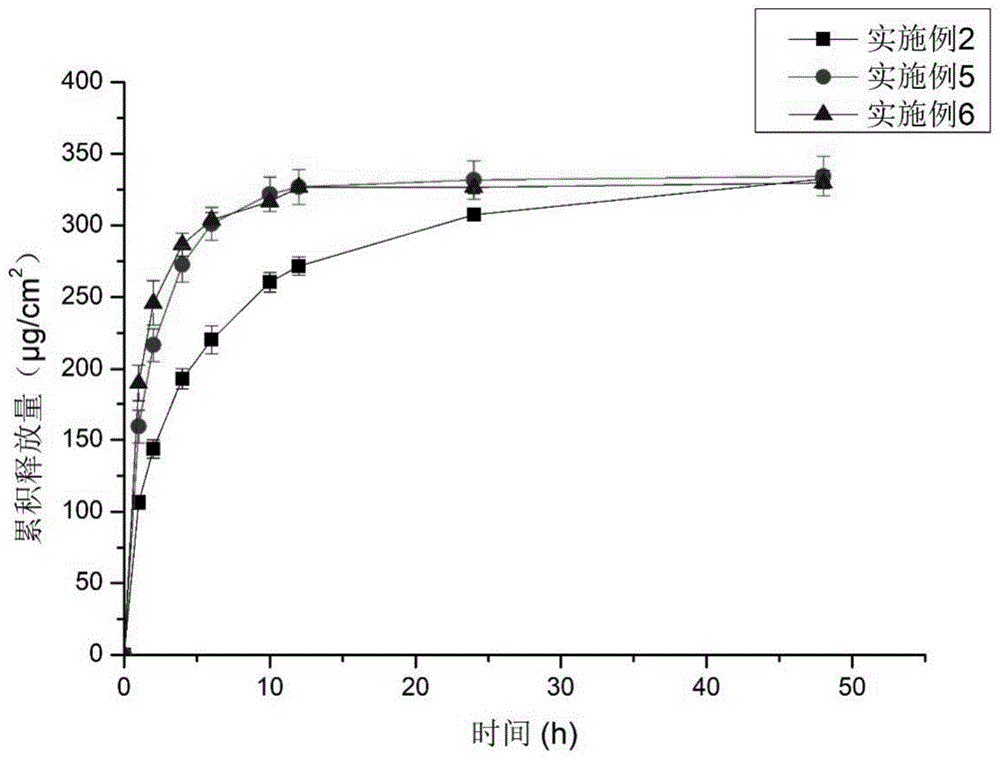
图1为实施例2、5、6的体外释放研究,考察加入聚丙烯酸树脂后对释放的影响。
图2为实施例4、7、8的大鼠皮肤体外透过实验,考察不同药物浓度对体外透过的影响
图3为实施例1、2、4分别制得的不加促渗剂和e100的贴剂、加促渗剂不加e100贴剂、既含促渗剂又含e100的贴剂,进行人皮体外透过实验。
图4为实施例4的大鼠药代动力学研究,将实施例4贴剂与市售注射剂进行药时曲线对比,确定该贴剂经皮给药的可行性。
其中:图1为实施例2、5、6的累积释放量-时间曲线(平均值±s.d,n=3)。
图2为实施例4、7、8的累积透过率-时间曲线(平均值±s.d,n=3)。
图3为实施例1、2、4的人皮累积透过率-时间曲线(平均值±s.d,n=3)。
图4为大鼠血浆药物浓度-时间曲线(平均值±s.d,n=3)。
在本发明中,所述的聚丙烯酸树脂为e100(德国罗姆公司生产、商品牌号尤特奇)和/或rlpo(德国罗姆公司生产、商品牌号尤特奇);所述的化学促渗剂为ipm(英国croda公司、crodamolipm-lq-(sg)),所述的抗氧剂为维生素e,所述的压敏胶可以为聚丙烯酸压敏胶(美国国民淀粉公司、dt-2287)。其中:e100的主成分为甲基丙烯酸丁酯、甲基丙烯酸二甲胺基乙酯和甲基丙烯酸甲酯(1:2:1)共聚物,rlpo主成分为丙烯酸乙酯、甲基丙烯酸甲酯和甲基丙烯酸氯化三甲胺基乙酯(1:2:0.2)。
具体实施方式:
以下结合实施例对本发明公开的技术方案做进一步说明,需要指出的是,以下说明仅仅是对本发明要求保护的技术方案的举例说明,并非对这些技术方案的任何限制。
在本发明中,如非特指,所有的量、份均为质量单位,所有的原料及生产设备均可以从市场购得。
实施例1
将0.1g(5%w/w)帕洛诺司琼碱溶解在5.278g聚丙烯酸酯压敏胶中,加入0.002g维生素e,加入2ml乙酸乙酯混匀,室温静置1h脱气,转移涂布在防粘层上,烘干过程溶剂,复合上背衬层,形成帕洛诺司琼透皮贴剂。
实施例2
将0.1g帕洛诺司琼碱溶解在4.722g聚丙烯酸酯压敏胶中,加入0.2gipm和0.002g维生素e,加入2ml乙酸乙酯混匀,室温静置1h脱气,转移涂布在防粘层上,烘干过程溶剂,复合上背衬层,形成帕洛诺司琼透皮贴剂。
实施例3
将0.2ge100用乙酸乙酯溶胀,加入4.167g聚丙烯酸压敏胶,形成均匀的压敏胶基质后,将0.1g帕洛诺司琼碱溶解在基质中,加入0.2gipm和0.002gve,加入2ml乙酸乙酯混合均匀,室温静置1h脱气,转移涂布在防粘层上,烘干过程溶剂,复合上背衬层,形成帕洛诺司琼透皮贴剂。
实施例4
将0.4ge100用乙酸乙酯溶胀,加入3.611g聚丙烯酸压敏胶,形成均匀的压敏胶基质后,将0.1g帕洛诺司琼碱溶解在基质中,加入0.2gipm和0.002gve,加入2ml乙酸乙酯混合均匀,室温静置1h脱气,转移涂布在防粘层上,烘干过程溶剂,复合上背衬层,形成帕洛诺司琼透皮贴剂。
实施例5
将0.6ge100用乙酸乙酯溶胀,加入3.179g聚丙烯酸压敏胶,形成均匀的压敏胶基质后,将0.1g帕洛诺司琼碱溶解在基质中,加入0.2gipm和0.002gve,加入2ml乙酸乙酯混合均匀,室温静置1h脱气,转移涂布在防粘层上,烘干过程溶剂,复合上背衬层,形成帕洛诺司琼透皮贴剂。
实施例6
将0.6grlpo用乙酸乙酯溶胀,加入3.179g聚丙烯酸压敏胶,形成均匀的压敏胶基质后,将0.1g帕洛诺司琼碱溶解在基质中,加入0.2gipm和0.002gve,加入2ml乙酸乙酯混合均匀,室温静置1h脱气,转移涂布在防粘层上,烘干过程溶剂,复合上背衬层,形成帕洛诺司琼透皮贴剂。
实施例7
将0.4ge100用乙酸乙酯溶胀,加入3.833g聚丙烯酸压敏胶,形成均匀的压敏胶基质后,将0.02g(1%w/w)帕洛诺司琼碱溶解在基质中,加入0.2gipm和0.002gve,加入2ml乙酸乙酯混合均匀,室温静置1h脱气,转移涂布在防粘层上,烘干过程溶剂,复合上背衬层,形成帕洛诺司琼透皮贴剂。
实施例8
将0.4ge100用乙酸乙酯溶胀,加入3.722g聚丙烯酸压敏胶,形成均匀的压敏胶基质后,将0.06g(3%w/w)帕洛诺司琼碱溶解在基质中,加入0.2gipm和0.002gve,加入2ml乙酸乙酯混合均匀,室温静置1h脱气,转移涂布在防粘层上,烘干过程溶剂,复合上背衬层,形成帕洛诺司琼透皮贴剂。
实施例2、5、6贴片进行影响因素实验
分别凿取2cm2的贴片放于行高温(60℃±2℃)、高湿(90%±5%rh)、高光(4500lx±500lx)实验,一式三份分别在0天、5天、10天检测单一未知杂质含量。
表1加入不同类型丙烯酸树脂贴片的影响因素实验(平均值±s.d,n=3)
结论:由以上实验发现,加入e100和rlpo均可降低该未知杂质(初步研究该杂质的产生与压敏胶的某些成分有关,具体还需进一步研究)含量,加入e100的贴剂药物稳定性要略好于加入rlpo的贴剂。
实施例2、5、6的体外释放实验
取2、5、6贴剂和横式扩散池,将贴片裁成合适大小(1.766cm2),用透明胶带将贴片固定在扩散池池口,加入接收介质(12ml、5%的乙醇-水),保持水浴循环温度为32±0.5℃,磁力搅拌转速为600r·min-1,取样时间点为1h、2h、4h、6h、10h、12h、24h、48h,每次从取样口取出200μl接收液,并补充200μl等温的新鲜介质。一式三份并通过hplc检测药物的含量,计算累积透过量。
结果如图1所示,加入e100/rlpo后能提高药物的释放速率,实施例5、6在12h释放完全,而未加聚丙烯酸树脂(实施例2)的贴片48h基本释放完全。
实施例2-5贴片的大鼠体外透过实验
(1)离体皮肤处理标准体重的sd雄性大鼠(200±20g)适应性喂养3天,用乙醚麻醉致死,用刀片将背部毛发剃刮干净(避免划伤角质层),剪下该处皮肤,去除皮下脂肪及结缔组织,选取角质层完整且厚度相近的皮肤,剪成2.0cm×2.0cm大小,储存在-20℃冰箱中以备用(一周内使用)。实验时将皮肤从冰箱中取出,恢复至室温,供体外皮肤渗透实验使用。
(2)体外经皮渗透实验方法取实施例2-5贴剂和横式扩散池,先将贴剂贴在角质层侧,位置对准扩散池的池口,皮肤的真皮侧面向接收池(体积为12ml、5%的乙醇-水为介质)用透气的胶带将皮肤固定在扩散池上,保持水浴循环温度为32±0.5℃,磁力搅拌转速为600r·min-1,取样时间点为1h、2h、4h、6h、10h、12h、24h、48h,每次从取样口取出200μl接收液,并补充200μl等温的新鲜介质。一式三份并通过hplc检测药物的含量,计算累积透过率。
表2贴片中加入不同比例e100的体外透过实验结果(平均值±s.d,n=3)
结论:加入不同比例e100,均能提高其累积透过率,加入0.4g达到最高累积透过率,再增加e100的比例并不能提高其透过率,因此0.4g(在贴片的中占胶层的20%)e100在贴片中为最佳比例。
实施例4、7、8的大鼠体外透过实验
实验方法如上所述,结果如图2所示,可以发现累积透过率是随药物浓度增加而增加,但在72h累积透过率均在78%以上,贴剂中药物浓度低可以通过增大给药面积来满足给药要求。
实施例1,2,4的人皮透过实验
采用与大鼠体外透过相同的实验方法,将大鼠皮肤更换人体皮肤,裁成合适大小,进行体外透过实验。结果如图3所示,实施例1和实施例2的对比说明ipm促进药物的经皮透过;实施例2和实施例4对比说明e100也有促进药物经皮透过的作用。因此本发明中ipm和e100共同促进药物经皮透过,能够实现给药1~3天能维持5~7天的药效。
实施例4的大鼠药代动力学实验
(1)实验动物分组及给药将6只sd大鼠(250±20g,雄性)随机分为注射组和贴剂组,每组3只。
注射组:按照每只给药20μg·kg-1的剂量尾静脉注射,于给药后0.0167、0.0833、0.25、0.5、1、2、4、6、10、12、24、48、72h自眼后静脉丛取血,取血量约为0.3ml置于含肝素钠润湿的ep管中。
贴剂组:提前24h将大鼠背部毛发用剃须刀刮干净(约为2×2cm2),于第二天进行实验,每只鼠按照84μg·kg-1给药。用模具凿取f2贴剂0.07cm2(含药量约300μg·cm-2)贴于剃毛且无损伤的皮肤处,并用透气的医用胶带固定贴剂48h,分别于给药后2、4、6、8、10、12、24、48、72h自眼后静脉丛取血。将以上两组取出的血液样品立即以3500r·min-1离心10min,分取上层血浆,储存在-40℃冰箱中保存。
(2)血浆样品处理将血浆样品从冰箱中取出,解冻至室温,取50μl血浆于ep管中,加入50μl饱和碳酸氢钠溶液和盐酸格拉司琼溶液(内标,5ng·ml-1)10μl,加1ml乙酸乙酯,充分涡旋,以12000r·min-1离心5min,取上清液,氮气吹干,50μl乙腈复溶,涡旋,离心,进行样品检测。
(3)lc-ms/ms条件色谱条件:用acquityuplcbehc18柱(50×2.1mm,1.7μm),流动相为20mmol·l-1甲酸铵(0.1%甲酸)溶液:甲醇(45:55),柱温40℃,流速0.2ml·min-1,进样量1μl。帕洛诺司琼保留时间在1.89min,格拉司琼保留时间在1.17min。
质谱条件:esi离子源,正离子方式检测;毛细管电压2.2kv;锥孔电压16v;离子源温度400℃;源抵消电压55v;雾化气流量1000l·h-1;碰撞气流量0.15ml·min-1。用于定量的离子反应:监测离子帕洛诺司琼:m/z297.2→110.1,格拉司琼:m/z313.3→138.0。
实验结果如图4和表3所示,注射组与贴剂组有明显不同的药时曲线,贴剂组的相对生物利用度为33.1%±13.2%。注射组在给药之初达到最大血药浓度,约为3.39ng·ml-1,随后迅速消除,消除半衰期为16.4±5.9h,血药浓度波动大;与之相对,贴剂组起初药物浓度低,大约6h左右达到最高血药浓度,约为0.46ng·ml-1,随后缓慢消除,消除半衰期达26.4±6.8h,维持了较长时间的有效血药浓度,且血药浓度波动范围较小。
- 还没有人留言评论。精彩留言会获得点赞!