一种环境友好型路线制备氮掺杂分级多孔碳材料及提高其比电容的方法与流程
本发明涉及电化学储能领域,具体涉及一种环境友好型路线制备氮掺杂分级多孔碳材料及提高其比电容的方法。
背景技术:
目前我国正大力发展清洁能源,然而清洁能源(如太阳能和风能)由于不稳定、连续性不好等缺点阻碍了其大规模应用,因此发展例如电池、电容器这些电化学储能被认为是解决清洁能源发电入网的有效途径。在这些储能设备中,电极材料是最为关键的组成部分,不仅决定了储能设备的性能,更影响价格。由于良好的导电性、价格低廉、制备工艺成熟等优点,碳材料成为应用最广泛的电极材料。为了进一步提升碳材料的性能,杂原子(尤其是氮原子)掺杂由于引入了电化学活性位点,已被证明是提高比电容有效的手段(j.mater.chem.a,2016,4(4):1144-1173;adv.sci.,2017:1600408)。另外,构建分级多孔结构,既可以获得较大比表面积,又可以达到离子快速传输的目的。因此,制备氮掺杂分级多孔碳材料是目前热门的研究内容。
制备氮掺杂分级多孔碳材料的方法有很多,其中常用的是煅烧含氮前驱体,前驱体中常常加入活化剂得到分级多孔结构。然而文献资料表明,利用碱性活化剂制备氮掺杂分级多孔碳材料会出现诸如氰化钾这样的剧毒物质(carbon,2019,144:745-755;carbon,2018,131:193-200)。由于煅烧后通常加入酸去除碱性活化剂煅烧后的残留杂质,若在煅烧过程中生成氰化钾,遇酸会生成氢氰酸这样挥发性的剧毒物质,对操作者的人身安全形成巨大威胁。
技术实现要素:
针对现有碱性活化剂制备氮掺杂分级多孔碳材料过程中会产生剧毒物质的不足,本发明的目的在于提供一种环境友好型路线制备氮掺杂分级多孔碳材料及提高其比电容的方法。
本发明提供了一种环境友好型路线制备氮掺杂分级多孔碳材料及提高其比电容的方法。该制备方法主要克服了传统制备路线中使用碱性活化剂制备氮掺杂多孔碳材料过程中产生剧毒物质(如kcn)的缺点,实现低污染的环保制备路线。相比使用碱性活化剂的路线,该制备方法得到的氮掺杂多孔碳材料具有更合理的多级孔结构和更高的氮含量,更适合作为高性能超级电容器的电极材料。同时,采用调变电解液的ph,可以大大提高材料的点电容。
本发明的目的至少通过如下技术方案之一实现。
本发明提供的一种环境友好型路线制备氮掺杂分级多孔碳材料的方法,包括如下步骤:
(1)将邻苯二甲酸、邻苯二甲酸氢钾和氯化钙加入水中,混合均匀,然后进行预冷处理,滴加乙二胺,搅拌均匀,得到混合溶液;
(2)将所述混合溶液进行冷冻干燥处理,得到干燥后的产物;
(3)在保护气氛下将步骤(2)所述干燥后的产物升温,进行煅烧处理,酸洗,水洗,干燥,得到所述氮掺杂分级多孔碳材料。
进一步地,步骤(1)所述邻苯二甲酸与邻苯二甲酸氢钾的摩尔比为1:1。
进一步地,步骤(1)所述邻苯二甲酸氢钾与乙二胺的摩尔比为1:3。
进一步地,步骤(1)所述氯化钙与邻苯二甲酸氢的钾摩尔比为0.5:1-2:1。
进一步地,步骤(1)所述预冷处理的温度为4℃,预冷处理的时间为15min。
进一步地,步骤(2)所述冷冻干燥处理的温度为-50℃,冷冻干燥处理的时间为12小时。
进一步地,步骤(3)所述保护气氛为氩气或氮气气氛;所述煅烧处理的温度为800℃,煅烧处理的时间为2小时;所述干燥的温度为80℃,干燥的时间为12小时。
本发明提供一种由上述的制备方法制得的氮掺杂分级多孔碳材料。
本发明提供的氮掺杂分级多孔碳材料在制备电容器中的应用,包括如下步骤:将所述氮掺杂分级多孔碳材料制成电极材料;然后将li2so4溶液作为电解液,往所述li2so4溶液中加入酸或碱,调节所述li2so4溶液的ph为2.5或ph为12.0,将所述电极材料和电解液组装成电容器。
进一步地,在所述的氮掺杂分级多孔碳材料在制备电容器中的应用中,所述li2so4溶液的浓度为1m;所述酸为h2so4溶液,所述碱为lioh。
本发明提供的制备方法,加入氯化钙等物质有效避免碱性活化剂制备氮掺杂分级多孔碳材料中的有毒物质,实现环境友好的制备路线,而且改进后流程制备出来的氮掺杂多孔碳材料相比原来氮含量更高,孔结构更合理,更适合作为高性能超级电容器电极材料。
本发明提供了一种提升所述氮掺杂分级多孔碳材料的比电容的方法,通过对电解液ph的调节,能够有效提高材料的比电容,提高材料的能量密度,推进其实际应用进程。
与现有技术相比,本发明具有如下优点和有益效果:
本发明提供的制备方法煅烧产物中克服了传统制备路线中使用碱性活化剂制备氮掺杂多孔碳材料过程中产生剧毒物质的缺点,实现低污染的环保制备路线。相比原来只使用碱性活化剂的路线,该方法制备得到的氮掺杂多孔碳材料具有更合理的分级孔结构和更高的氮含量,更适合作为高性能超级电容器的电极材料。提高材料比电容的方法操作简单,效果明显且环境友好。
附图说明
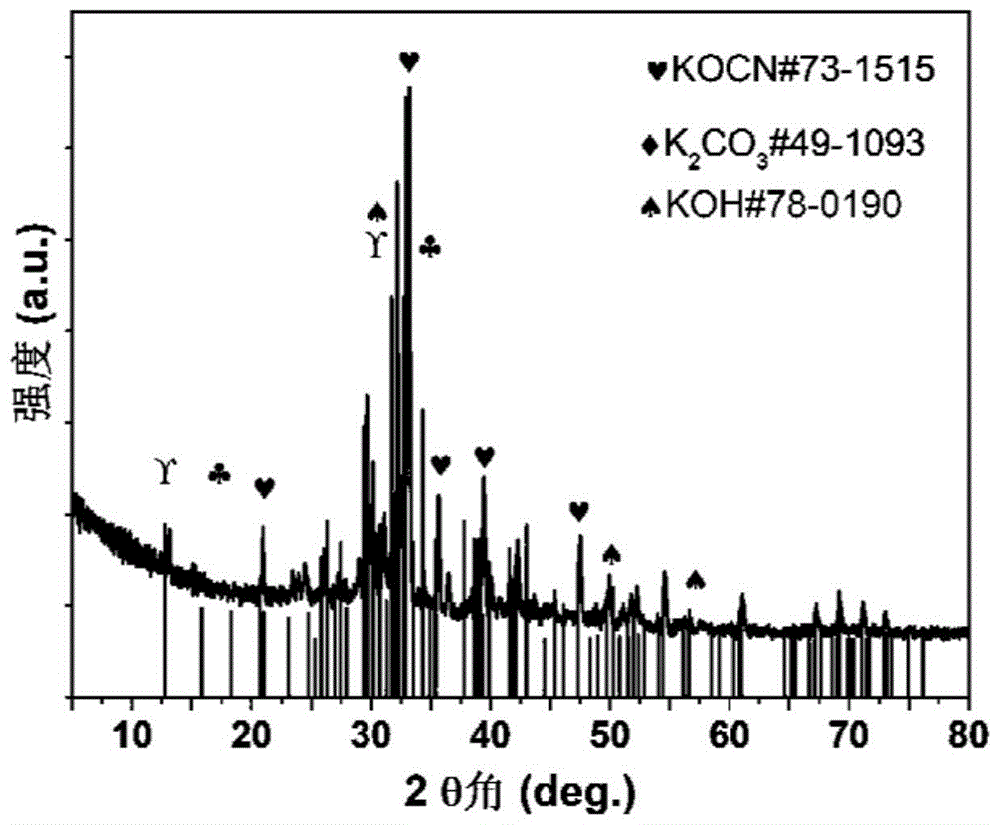
图1为对比例1制备的材料煅烧后酸洗前的xrd图;
图2为实施例1制备的材料煅烧后酸洗前的xrd图;
图3为实施例2制备的材料煅烧后酸洗前的xrd图;
图4为实施例3制备的材料煅烧后酸洗前的xrd图;
图5为实施例1和对比例1制备的材料的xrd图;
图6为实施例1和对比例1制备的材料的氮气吸脱附曲线图和孔径分布图;
图7为实施例1和对比例1制备的碳基材料在6mkoh三电极体系下电流密度-比容量关系曲线;
图8为实施例1得到的碳基材料在6mkoh溶液三电极体系10ag-1电流密度5000次恒电流充放电长循环结果图。
图9为实施例1得到的碳基材料在实施例4、实施例5和对比例2中测试结果图。
具体实施方式
以下结合实例对本发明的具体实施作进一步说明,但本发明的实施和保护不限于此。需指出的是,以下若有未特别详细说明之过程,均是本领域技术人员可参照现有技术实现或理解的。所用试剂或仪器未注明生产厂商者,视为可以通过市售购买得到的常规产品。
实施例1
一种环境友好型路线制备氮掺杂分级多孔碳材料的方法,包括如下步骤:
(1)将邻苯二甲酸(3.3226g,0.02mol)、邻苯二甲酸氢钾(4.0844g,0.02mol)和氯化钙(1.1099g,0.01mol)置于100ml烧杯中,加入40ml去离子水中,混合均匀,得到悬浊液,然后将所述悬浊液置于4℃环境中进行预冷处理,预冷处理的时间为15min,滴加3.6060g(0.06mol)乙二胺,搅拌均匀,搅拌时间为10min,得到混合溶液;
(2)将所述混合溶液进行冷冻干燥处理,冷冻干燥处理的温度为-50℃,冷冻干燥处理的时间为12h,得到干燥后的产物(前驱体);
(3)将步骤(2)所述干燥后的产物(前驱体)研磨成粉后,在氮气气氛下将所述干燥后的产物(前驱体)升温,升温速率为5℃/min,进行煅烧处理,煅烧处理的温度为800℃,煅烧处理的时间为2h,冷却至室温,得到碳化物,将所述碳化物加入200ml浓度为0.2mol/l的盐酸溶液中进行酸洗,搅拌12h,过滤取沉淀,得到酸洗后的碳材料,水洗所述酸洗后的碳材料至滤液呈中性,抽滤取沉淀,干燥,干燥的温度为80℃,干燥的时间为12h,得到所述氮掺杂分级多孔碳材料。
实施例2
一种环境友好型路线制备氮掺杂分级多孔碳材料的方法,包括如下步骤:
(1)将邻苯二甲酸(3.3226g,0.02mol)、邻苯二甲酸氢钾(4.0844g,0.02mol)和氯化钙(2.2198g,0.02mol)置于100ml烧杯中,加入40ml去离子水中,混合均匀,得到悬浊液,然后将所述悬浊液置于4℃环境中进行预冷处理,预冷处理的时间为15min,滴加3.6060g(0.06mol)乙二胺,搅拌均匀,搅拌时间为10min,得到混合溶液;
(2)将所述混合溶液进行冷冻干燥处理,冷冻干燥处理的温度为-50℃,冷冻干燥处理的时间为12h,得到干燥后的产物(前驱体);
(3)将步骤(2)所述干燥后的产物(前驱体)研磨成粉后,在氮气气氛下将所述干燥后的产物(前驱体)升温,升温速率为5℃/min,进行煅烧处理,煅烧处理的温度为800℃,煅烧处理的时间为2h,冷却至室温,得到碳化物,将所述碳化物加入200ml浓度为0.2mol/l的盐酸溶液中进行酸洗,搅拌12h,过滤取沉淀,得到酸洗后的碳材料,水洗所述酸洗后的碳材料至滤液呈中性,抽滤取沉淀,干燥,干燥的温度为80℃,干燥的时间为12h,得到所述氮掺杂分级多孔碳材料。
实施例3
一种环境友好型路线制备氮掺杂分级多孔碳材料的方法,包括如下步骤:
(1)将邻苯二甲酸(3.3226g,0.02mol)、邻苯二甲酸氢钾(4.0844g,0.02mol)和氯化钙(4.4396g,0.04mol)置于100ml烧杯中,加入40ml去离子水中,混合均匀,得到悬浊液,然后将所述悬浊液置于4℃环境中进行预冷处理,预冷处理的时间为15min,滴加3.6060g(0.06mol)乙二胺,搅拌均匀,搅拌时间为10min,得到混合溶液;
(2)将所述混合溶液进行冷冻干燥处理,冷冻干燥处理的温度为-50℃,冷冻干燥处理的时间为12h,得到干燥后的产物(前驱体);
(3)将步骤(2)所述干燥后的产物(前驱体)研磨成粉后,在氮气气氛下将所述干燥后的产物(前驱体)升温,升温速率为5℃/min,进行煅烧处理,煅烧处理的温度为800℃,煅烧处理的时间为2h,冷却至室温,得到碳化物,将所述碳化物加入200ml浓度为0.2mol/l的盐酸溶液中进行酸洗,搅拌12h,过滤取沉淀,得到酸洗后的碳材料,水洗所述酸洗后的碳材料至滤液呈中性,抽滤取沉淀,干燥,干燥的温度为80℃,干燥的时间为12h,得到所述氮掺杂分级多孔碳材料。
实施例4
以1.0mli2so4溶液为电解液主体,加入2mh2so4溶液调节所述li2so4溶液的ph至2.5,作为电解液与实施例1制得的氮掺杂分级多孔碳材料组装成三电极体系,然后测试实施例1中得到的氮掺杂分级多孔碳材料的电化学性能。
实施例5
以1.0mli2so4溶液为主体,加入1.0mlioh溶液调节溶液的ph至12作为电解液,作为电解液与实施例1制得的氮掺杂分级多孔碳材料组装成三电极体系,然后测试实施例1中得到的氮掺杂分级多孔碳材料的电化学性能。
对比例1
(1)将邻苯二甲酸(3.3226g,0.02mol)和邻苯二甲酸氢钾(4.0844g,0.02mol)置于100ml烧杯中,加入40ml去离子水,搅拌得悬浊液;
(2)将步骤(1)得到的悬浊液置于4℃环境中预冷15min,然后加入3.6060g(0.06mol)乙二胺,搅拌均匀,搅拌的时间为10min以充分混合,得到澄清溶液;
(3)将步骤(2)得到的澄清溶液放在冷冻干燥机中以-50℃冷冻干燥12h后得到前驱体;
(4)将前驱体研磨成粉后,在氮气气氛中保护下,以5℃/min的升温速度升至800℃煅烧2h,冷却至室温,得到碳化物。将所得碳化物加入体积为200ml的盐酸溶液(浓度为0.2mol/l)中进行酸洗,搅拌12h,过滤取沉淀,得到酸洗后的碳材料,用去离子水洗涤至中性,将抽滤后的材料在80℃干燥12h,即得到氮掺杂多孔碳材料。
对比例2
采用1.0mli2so4溶液(ph为7.0)作为电解液与实施例1制得的氮掺杂分级多孔碳材料组装成三电极体系,然后测试实施例1中得到的碳基材料的电化学性能。
对实施例1、实施例2、实施例3和对比例1煅烧后酸洗前的碳化物采用x射线粉末衍射仪(德国布鲁克d8advance)定性检测其中组分。扫描角度为10-80°,并且与标准卡片比对得到它们的组成,如图1、图2、图3和图4所示。首先对比例1与文献比较,粗产物并未发现剧毒物质仅有低毒的氰酸钾;其次加入不同比例的氯化钙后粗产物中并未发现氰酸钾,体现了对环境友好制备路线的特点。
对实施例1和对比例1干燥后得到的碳材料采用x射线粉末衍射仪(德国布鲁克d8advance)测试其晶体结构,扫描角度为10-80°,可知样品在25°附近具有碳材料特有的包峰,见图5所示。可发现两种材料晶体结构类似,表明实施例提供的制备方法的改变并未对氮掺杂碳材料的晶体结构产生明显影响。
对实施例1和对比例1干燥后得到的碳材料利用比表面分析仪(北京贝士德3h-2000ps1)测试其比表面积,得到氮气吸附脱曲线和孔径分布,如图6。可以看到实施例1相比对比例1微孔体积有所下降,但是介孔和大孔体积明显上升,调整了孔道结构,有利于离子快速迁移。
将实施例1和对比例1干燥后得到的碳材料制成电极材料,在电化学工作站(上海辰华chi660e)组装成三电极体系,其中参比电极为hg/hgo,对电极为1cm×1cm铂网电极,电解液为6mkoh,在不同电流密度(0.5、1.0、2.0、3.0、5.0、10、20、30、50和100ag-1)下的充放电性能,并通过放电时间计算样品在不同电流密度下的比电容,结果如图7所示。可看到对比例1和实施例1容量保持率相近,但实施例1得到的碳基材料在任意电流密度下的比电容均高于对比例1获得的碳材料,表明该制备方法得到的材料更适合作为高性能超级电容器电极材料。
将实施例1得到的碳基材料在6mkoh溶液中三电极体系下以10ag-1电流密度进行5000次长循环,结果如图8所示。循环保持率达到100.3%,几乎没有衰减,体现良好的电化学稳定性。
将实施例1得到的碳基材料在实施例4、实施例5和对比例2环境中三电极体系下测试,测试结果如图9所示。由图9可知实施例4和实施例5在任意电流密度下容量均高于对比例2,表明该提高比电容的方法有效且效果明显。
表1是实施例1和对比例1材料氮含量、比表面积、平均孔径、总孔体积和微孔体积的测试结果。由表1可得,实施例1氮含量相比对比例1更高。
表1
以上实施例仅为本发明较优的实施方式,仅用于解释本发明,而非限制本发明,本领域技术人员在未脱离本发明精神实质下所作的改变、替换、修饰等均应属于本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!