一种阳离子型可见光固化组合物的制作方法
本发明属于光聚合技术领域,具体涉及一种阳离子型可见光固化组合物。
背景技术:
双酚A型环氧树脂是热固性树脂中固化收缩率最小的一种,其本身具有很高的粘合力,其固化制成品具有优异的机械性能、耐化学品性能和电绝缘性能,且价格低廉,广泛应用于胶黏剂、快速成型等多种领域。传统工艺主要采用热固化成型技术对其进行固化,但工艺复杂、成本高。
随着环氧树脂光辐射固化成型技术的发展,对双酚A型环氧树脂的光固化研究也有了一些进展,主要集中在双酚A型环氧树脂的紫外光固化体系中。但紫外光源对设备有较高要求,辐射大、环保安全性差,且固化成本高,极大地限制了其应用。
因此,开发更加安全、成本低、污染小的双酚A型环氧树脂可见光固化体系具有研究价值和实际意义。
技术实现要素:
为了克服上述问题,本发明人对光固化技术进行了锐意研究,制备出一种阳离子型可见光固化组合物,其采用特定结构和含量的阳离子引发剂配合增感剂,能够在可见光的照射下引发双酚A型环氧树脂固化成型。
本发明的目的在于提供一种阳离子型可见光固化组合物,其包括以下重量配比的组分:
引发剂 0.5~10重量份;
活性稀释剂 0.5~30重量份;和
双酚A型环氧树脂 70~90重量份,
其中,所述引发剂包括阳离子光引发剂和增感剂。
所述阳离子光引发剂和增感剂的重量比为1:(0.01~1),优选为1:(0.05~0.8),更优选为1:(0.25~0.5)。
所述阳离子光引发剂为如式(Ⅰ)所示的二芳基碘鎓盐。
其中,R1选自-H、-R’和-OR’,R’为C1-12的烷基;
X-选自SbF6-,AsF6-,PF6-和BF4-。
所述增感剂具有如式(Ⅱ)所示结构。
其中,R2、R3、R4选自-H、-R”、-OR”和-Br,且R2、R3、R4中至少有一个为-Br,R”为C1-12的烷基。
所述双酚A型环氧树脂的环氧值为0.35~0.57,优选为0.41~0.52。
如上所述的组合物,其包括以下重量配比的组分:
所述活性稀释剂选自环氧氯代烷、氯代烷、酮、和芳香杂环化合物中的一种或几种。
所述组合物在可见光源照射下发生聚合反应,固化成型。
所述可见光源的波长为380~600nm。
本发明所具有的有益效果包括:
(1)本发明提供的阳离子型可见光固化组合物能够在可见光的照射下固化成型,对设备要求低,成本更低,安全性更高且对环境污染更小;
(2)本发明提供的阳离子型可见光固化组合物的光感性强,且光固化转化率高,在小于30mW/cm2的低照度下其固化转化率可达50%;
(3)本发明提供的阳离子型可见光固化组合物中采用特定的阳离子光引发剂和增感剂作为光固化反应的引发剂,其引发速度快、效率高,具有良好的稳定性,毒性低、对环境无污染,且价廉易得,成本低。
附图说明
图1示出实验例1测得的紫外-可见吸收光谱图;
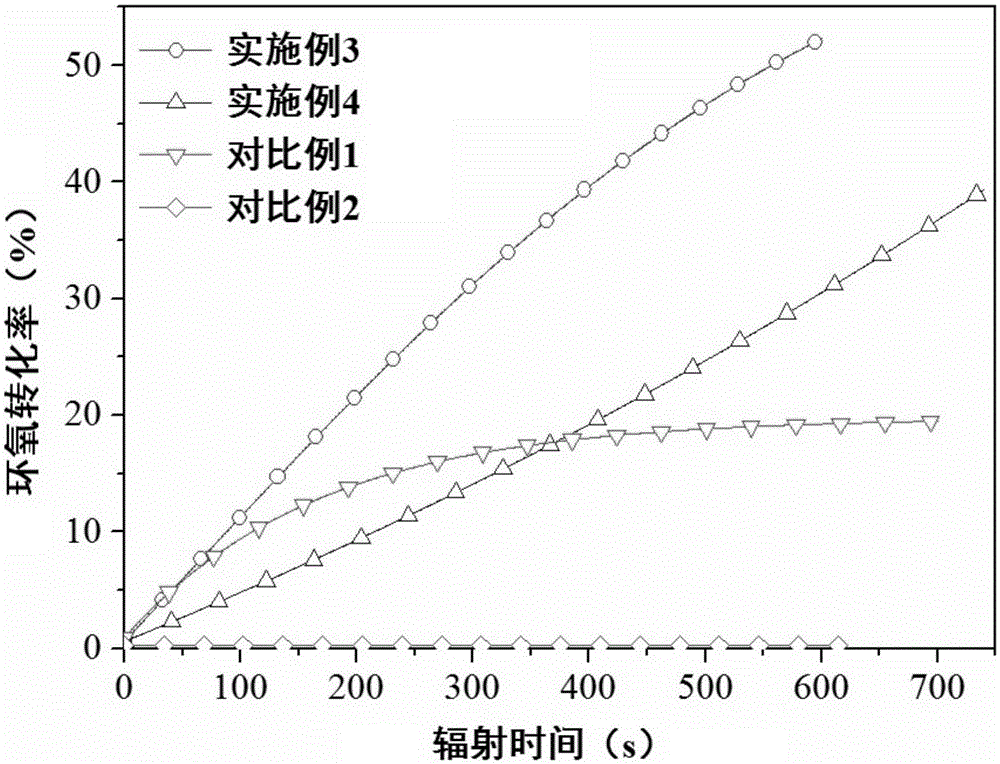
图2示出实验例2得到的环氧转化率随光照时间的变化曲线图;
图3示出实验例3得到的环氧转化率随光照时间的变化曲线图。
具体实施方式
下面通过附图、实验例和实施例对本发明进一步详细说明。通过这些说明,本发明的特点和优点将变得更为清楚明确。
根据本发明,提供一种阳离子型可见光固化组合物,其包括以下重量配比的组分:
引发剂 0.5~10重量份;
活性稀释剂 0.5~30重量份;和
双酚A型环氧树脂 70~90重量份,
所述引发剂包括阳离子光引发剂和增感剂,其中,所述阳离子光引发剂和增感剂的重量比为1:(0.01~1),优选为1:(0.05~0.8),更优选为1:(0.25~0.5)。
在根据本发明的优选实施方式中,所述阳离子光引发剂为如式(Ⅰ)所示的二芳基碘鎓盐,其能够在紫外光区吸收一定波长的能量,产生阳离子,从而引发环氧单体或环氧预聚体发生聚合反应交联固化,其最大吸收波长为250nm左右。
其中,R1选自-H、-R’和-OR’,R’为C1-12的烷基,优选地,R1选自-H和C1-6的烷基。
X-选自SbF6-,AsF6-,PF6-和BF4-,优选为PF6-或BF4-。
更优选地,所述阳离子光引发剂为二苯基碘鎓六氟磷酸盐(如式(Ⅲ)所示)、4,4’-二甲基二苯基碘鎓六氟磷酸盐、二苯基碘鎓四氟硼酸盐或4,4’-二甲基二苯基碘鎓四氟硼酸盐。
上述引发剂为已知化合物,可以商购获得,也可以根据现有方法合成得到,在此不作赘述。
在根据本发明的优选实施方式中,所述增感剂为如式(Ⅱ)所示的BODIPY(氟化硼二吡咯)类荧光染料,其在可见光区具有较强吸收,最大吸收波长为540nm左右,上述增感剂与如式(Ⅰ)所示的阳离子光引发剂按照特定比例混合后,可使感光范围扩大至整个可见光区域,从而能够利用可见光来引发环氧单体或环氧预聚体的交联固化反应。
其中,R2、R3、R4选自-H、-R”、-OR”和-Br,且R2、R3、R4中至少有一个为-Br,R”为C1-12的烷基,优选为C1-6的烷基。
更优选地,所述增感剂为如式(Ⅳ)或式(Ⅴ)所示的BODIPY类荧光染料。
本发明中所用增感剂,可以参照已知方法合成获得,所述方法参见RSC Advances,2012,2,9851-9859。
所述双酚A型环氧树脂具有如式(Ⅵ)所示结构,其环氧值为0.35~0.57,优选为0.41~0.52。
其中,n为≥0的自然数。
所述双酚A型环氧树脂优选为双酚A型环氧树脂E51。
在根据本发明的优选实施方式中,所述活性稀释剂选自环氧氯代烷、氯代烷、酮、和芳香杂环化合物中的一种或几种,其中,所述环氧氯代烷优选为环氧氯乙烷或环氧氯丙烷,所述氯代烷优选为二氯甲烷或三氯甲烷,所述酮优选为丙酮,所述芳香杂环化合物优选为四氢呋喃。
更优选地,所述活性稀释剂为环氧氯代烷,如环氧氯乙烷或环氧氯丙烷。
在根据本发明的优选实施方式中,所述阳离子型可见光固化组合物包括以下重量配比的组分:
优选地,所述组合物包括以下重量配比的组分:
在根据本发明的优选实施方式中,所述阳离子型可见光固化组合物的制备方法如下:
在避光条件下将所述阳离子光引发剂、增感剂和活性稀释剂混合,搅拌至引发剂完全溶解,然后加入双酚A型环氧树脂,搅拌均匀后避光保存。
本发明提供的所述阳离子型可见光固化组合物在波长为380~600nm的可见光源照射下可发生聚合反应,固化成型,且在小于30mW/cm2的低照度下其固化转化率可达50%。
所述可见光源可以为氙灯、镝灯、卤素灯、钨卤灯、LED灯、激光、自然光中的一种或多种,优选波长为450~550nm。
实施例及实验例中所用试剂、仪器来源如下:
双酚A型环氧预聚体E51,江苏三木集团;
卤素灯(50W,λ=380~580nm),苏州汇毅工业材料有限公司;
照度计,北京师范大学光电仪器厂;
红外光谱仪(5700),美国Nicolet公司。
实施例
实施例1 BODIPY染料-1的制备
制备如式(Ⅳ)所示的BODIPY染料-1(记为BPY-1):取500mL单口瓶,将2mmol吡咯和1mmol对溴苯甲醛溶解在200mL二氯甲烷中,用氩气置换烧瓶中的空气,并用氩气鼓泡的方法将溶液中的空气赶出,然后滴入一滴三氟乙酸作催化剂,在氩气保护下持续搅拌,反应温度为室温(25℃)。2小时后终止反应,用0.l mol/L的NaOH溶液100mL将反应液洗涤两遍,然后用无水硫酸钠干燥有机层,在氩气保护条件下将溶剂二氯甲烷全部蒸出,然后再加入20mL甲苯将蒸干的残渣溶解,l mmol2,3-二氯-5,6-二氰基苯醌作为氧化剂加入反应混合物中,10分钟后将8mmol三氟化硼乙醚溶液和8mmol三乙胺同时加入到反应体系中,持续搅拌2小时后采用硅胶柱分离,硅胶颗粒大小为200-300目,洗脱剂的配比为石油醚/正乙酸乙酯(体积比)=9:1,得到褐红色晶体,产率41%。
采用核磁共振氢谱(1H-NMR)、碳谱(13C-NMR)和飞行时间质谱(TOF-MS)对上述产物进行结构鉴定,结果如下:
1H-NMR(400MHz,CDCl3):δ=7.96(s,2H),7.69(d,J=8.4Hz,2H),7.45(d,J=8.4Hz,2H),6.91(d,J=4.1Hz,2H),6.56(d,J=3.7Hz,2H);
13C-NMR(101MHz,CDCl3):δ=145.77,144.59,134.70,132.63,131.84,131.32,125.52,118.83;
TOF-MS-ES+:347.0169,C15H10N2BBrF2分子量346.0088。
上述结果显示,按照本发明实施例1所述方法制备得到的产物为如式(Ⅳ)所示的BODIPY染料-1。
实施例2 BODIPY染料-2的制备
制备如式(Ⅴ)所示的BODIPY染料-2(记为BPY-2):取500mL单口瓶,将2mmol吡咯和1mmol对己氧基苯甲醛溶解在200mL二氯甲烷中,用氩气置换烧瓶中的空气,并用氩气鼓泡的方法将溶液中的空气赶出,然后滴入一滴三氟乙酸作催化剂,在氩气保护下持续搅拌,反应温度为室温(25℃)。2小时后终止反应,用0.l mol/L的NaOH溶液100mL将反应液洗涤两遍,然后用无水硫酸钠干燥有机层,在氩气保护条件下将溶剂二氯甲烷全部蒸出,然后再加入20mL甲苯将蒸干的残渣溶解,l mmol2,3-二氯-5,6-二氰基苯醌作为氧化剂加入反应混合物中,10分钟后将8mmol三氟化硼乙醚溶液和8mmol三乙胺同时加入到反应体系中,持续搅拌2小时后采用硅胶柱分离,硅胶颗粒大小为200-300目,洗脱剂的配比为二氯甲烷/正己烷(体积比)=1:2,得到桔红色固体中间体。取125mL单口瓶,将1.5mmol中间体加入CCl4(50mL)溶液中,然后再加入1.5mmol N-溴代丁二酰亚胺和0.1mmol过氧化苯甲酰,氮气保护、回流条件下强力搅拌6h。粗产品真空下旋转蒸发浓缩,硅胶柱分离提纯(环己烷/二氯甲烷(体积比)=5:1),得到黑红色固体,产率47%。
采用核磁共振氢谱(1H-NMR)、碳谱(13C-NMR)和飞行时间质谱(TOF-MS)对上述产物进行结构鉴定,结果如下:
1H-NMR(400MHz,CDCl3):δ=7.94(d,J=21.4Hz,1H),7.76(s,1H),7.52(dd,J=8.3,6.3Hz,2H),7.11–6.90(m,4H),6.64–6.51(m,1H),4.05(t,J=6.5Hz,2H),1.92–1.76(m,2H),1.59–1.28(m,6H),0.97–0.86(m,3H);
13C-NMR(101MHz,CDCl3):δ=162.16,145.25,143.30,141.34,132.91,132.51,130.11,125.59,119.22,114.76,114.55,105.64,68.45,31.55,29.10,25.70,22.60,14.03;
TOF-MS-ES+:449.1040,C21H22N2OBBrF2分子量446.0977。
上述结果显示,按照本发明实施例2所述方法制备得到的产物为如式(Ⅴ)所示的BODIPY染料-2。
实施例3 阳离子型可见光固化组合物的制备(一)
称取0.2000g(0.469mmol)二苯基碘鎓六氟磷酸盐(IPF),添加0.1000g(0.289mmol)实施例1制得的BODIPY染料-1(BPY-1)置于棕色瓶中,然后加入2mL环氧氯丙烷,充分搅拌使引发剂完全溶解,再加入10g双酚A型环氧预聚体E51,搅拌均匀,得到所述阳离子型可见光固化组合物,避光保存。
实施例4 阳离子型可见光固化组合物的制备(二)
按照类似于本发明实施例3中所述方法进行制备,区别仅在于:将BODIPY染料-1(BPY-1)替换为0.1288g(0.289mmol)BODIPY染料-2(BPY-2)。
对比例
对比例1
按照类似于本发明实施例3中所述方法进行制备,区别仅在于:不加入BODIPY染料-1(BPY-1),即不加入增感剂。
对比例2
按照类似于本发明实施例3中所述方法进行制备,区别仅在于:不加入二苯基碘鎓六氟磷酸盐(IPF),即不加入阳离子光引发剂。
实验例
实验例1
分别配制3×10-6mol/L的二苯基碘鎓六氟磷酸盐(IPF)、BODIPY染料-1(BPY-1)和BODIPY染料-2(BPY-2)的二氯甲烷溶液,使用紫外-可见吸收光谱仪测定上述三个样品的吸收光谱曲线,波长范围400~600nm,结果见图1。
如图1所示,IPF在400nm以上的可见光区无吸收;而BPY-1和BPY-2在可见光区具有较强吸收,其最大吸收峰在在510nm附近。
实验例2
将实施例3~4和对比例1~2制得的阳离子型可见光固化组合物样品均匀涂在两片玻璃之间的橡胶圈(直径固定)中,通过50W卤素灯(λ=380~580nm,照度25.0mW/cm2)在室温下辐照,每个样品通过近红外扫描重复实验三次。需要说明的是,同一样品点在卤素灯下每隔一段时间辐照一次立即进行一次红外扫描。
通过傅立叶变换近红外光谱监测双酚A型环氧预聚体中环氧基团在6072cm-1附近处的的特征峰随光照时间的变化,并按下式计算固化转化率(即:环氧转化率):
环氧转化率=[1-(St/Rt)/(S0/R0)]×100%
其中,St是光照时间t时所对应的环氧基团特征峰面积;
Rt是光照时间t时所对应的参比峰面积;
S0是t=0时所对应的环氧基团特征峰面积;
R0是t=0所对应的参比峰面积。
分别计算不同光照时间下的环氧转化率,绘制环氧转化率随光照时间的变化曲线,见图2。
如图2所示,在25.0mW/cm2照度卤素灯(λ=380~580nm)的照射下:
实施例3制得的组合物(2wt%IPF,1wt%BPY-1)可发生光聚合反应,固化成型,其环氧转化率达到50%;
实施例4制得的组合物(2wt%IPF,1wt%BPY-2)可发生光聚合反应,固化成型,其环氧转化率达到40%以上;
对比例1制得的组合物(2wt%IPF,不含增感剂)可发生光聚合反应,固化成型,其环氧转化率显著降低,不超过20%;
对比例2制得的组合物(1wt%BPY-1,不含阳离子光引发剂)不能发生光聚合反应。
实验例3
将实施例3~4和对比例1制得的阳离子型可见光固化组合物样品均匀涂在两片玻璃之间的橡胶圈(直径固定)中,通过50W卤素灯(λ=380~580nm,照度15.0mW/cm2)在放置窄带滤光片λ=540nm后于室温下辐照,每个样品通过近红外扫描重复实验三次。需要说明的是,同一样品点在卤素灯下每隔一段时间辐照一次立即进行一次红外扫描。
通过傅立叶变换近红外光谱监测双酚A型环氧预聚体中环氧基团在6072cm-1附近处的的特征峰随光照时间的变化,按实验例2中所述公式计算固化转化率(即:环氧转化率),并绘制环氧转化率随光照时间的变化曲线,见图3。
如图3所示,在15.0mW/cm2照度卤素灯(λ=540nm)的照射下:
实施例3制得的组合物(2wt%IPF,1wt%BPY-1)可发生光聚合反应,固化成型,其环氧转化率接近30%;
实施例4制得的组合物(2wt%IPF,1wt%BPY-2)可发生光聚合反应,固化成型,其环氧转化率达到15%以上;
对比例1制得的组合物(2wt%IPF,不含增感剂)不能发 生光聚合反应。
以上结合优选实施方式和范例性实例对本发明进行了详细说明。不过需要声明的是,这些具体实施方式仅是对本发明的阐述性解释,并不对本发明的保护范围构成任何限制。在不超出本发明精神和保护范围的情况下,可以对本发明技术内容及其实施方式进行各种改进、等价替换或修饰,这些均落入本发明的保护范围内。本发明的保护范围以所附权利要求为准。
- 还没有人留言评论。精彩留言会获得点赞!