用于制备放射性药物的试剂盒的制作方法
本发明涉及用于制备放射性药物的稳定的试剂盒。尤其,本发明涉及用于稳定试剂盒的配体组分的非水性溶剂的用途。
必须在有限的时间内制备和施用放射性药物,因为应用中使用的大部分放射性核素的短的半衰期。其通常由在GMP条件下产生的试剂盒配制。试剂盒通常包含待与放射性核素,比如99mTc络合的适用的配体;足够量的还原剂;调整pH以适合最佳标记条件的缓冲剂;稳定试剂;和赋形剂。以冻干或冷冻干燥的形式制备试剂盒,其增加了稳定性和货架期。试剂盒可容易运输并且储存,然后使用指定的放射性核素重构。冷冻干燥的试剂盒简化了标记并确保了用于标记的更稳定的条件。
冷冻干燥的试剂盒制剂的可用性有利于负责的医院人员容易制备用于施用的放射性药物,因为其仅仅涉及添加放射性核素和必要时的加热。所以,这些制备步骤在医院责任人员的能力范围内。
放射性药物的例子是99m锝-乙烯双半胱氨酸脱氧葡糖胺(99mTc-ECDG)1。99mTc-ECDG是单光子发射计算机断层成像术(SPECT)/计算机断层成像术(CT)(SPECT/CT)成像剂,其对于检测肺癌基本损害的能力,目前在美国处在三期临床试验1。99mTc-ECDG的成像能力与正电子成象术(PET)/CT成像剂,18F-氟脱氧葡萄糖(18F-FDG)相当2,其广泛用于(大于95%的扫描)检测冬眠心肌和代谢活性癌组织2。相对于18F-FDG潜在实施99mTc-ECDG的背后主要驱动力是相比PET放射性指示剂与采用SPECT放射性指示剂相关的显著更低的成本和在肺癌成像中实现相同水平的质量和效率相关的显著更低的成本3。
提出99mTc-ECDG的作用机制是经氨基己糖途径发生,原因是包含两个葡糖胺取代基。葡糖胺通过氨基己糖生物合成途径进入细胞并且其葡糖胺-6-磷酸酯的调节产物介导胰岛素活化下游和标志糖基化以及癌症生长2。在氨基己糖途径中,上调葡萄糖转运蛋白(glucose transporter)促进了谷氨酰胺:6-磷酸-果糖酰基转移酶(fructose-6-phosphate amidotransferase)(GFAT)的过表达。磷酸化的葡糖胺结合尿苷二磷酸(UDP)以形成UDPN-乙酰葡糖胺(UDP-GLcNAc)。通过O连接的蛋白质N-乙酰葡糖胺(O-GlcNAc)转移酶,细胞核和细胞溶质蛋白质上丝氨酸和苏氨酸残基的糖基化在所有多细胞真核生物中是常见的。糖基化是翻译后修饰的一部分并且好像修饰许多核质蛋白质。O-GlcNAc转移酶活性与细胞内UDP-GLcNAc和UDP浓度高度相关,其又与葡萄糖浓度和其他刺激高度相关。在细胞核内,普遍存在的转录因子Sp1被O-GlcNAc高度修饰。响应高血糖或升高的葡糖胺,Sp1经历高糖基化(hyperglycosylation)。因为O-GlcNAc参与氨基己糖途径和核活性,对于肿瘤的辨别诊断,其成为吸引人的成像剂。
对于公开的合成的文献调查(参考文献[5]、[6]、[7]和[8])总结了如何产生ECDG 3的数个实验方法。不幸的是,这些公开的程序都未成功可重现的(reproducible),因为这些合成涉及将ECDG暴露于水性介质,这证明了是无效的,因为ECDG已经显示对于空气、光水和温度敏感9。已经描述了ECDG的合成是一项具有挑战性的任务,原因是该配体非常易变的性质9。因为ECDG被期望用作成像剂,材料必须是药学级别的,这意味着需要进行纯化步骤,而基本上不实质损失产率。这证明是非常困难的,因为ECDG的低稳定性。
使99mTc EsCDG用作核医学环境中的放射性药物的问题复杂化的第二个因素是其在试剂盒制剂中的存在。
产生ECDG——一种水不稳定的配体——的试剂盒是存在问题的,因为一般的试剂盒程序包括在水相中的冻干步骤,其中纯的配体活性药学成分(API)溶于包含还原剂、添加剂和缓冲剂的每个的至少一种的水/生理盐水,分发在小瓶中并且被冷冻干燥。在医院,生理盐水中的99mTc被添加至试剂盒并且重构。然后,99mTc与ECDG配体螯合并且准备99mTc-ECDG放射性药物用于注射。发明人已经发现ECDG在水中几乎立即分解。仅仅当金属离子与ECDG,比如在99mTc-ECDG的情况下螯合时,其在水中是稳定的。
所以,仍需要包括稳定的组分的试剂盒系统,其允许适于诊断、治疗或其他指示应用的简单的、可重复和稳定的标记技术。此外,仍需要在管理认可的可接受的放射化学纯度水平并且同时保持高的稳定性、纯度和产率的有效的配体的放射性标记。
技术实现要素:
根据本发明的第一方面,提供用于制备放射性药物的试剂盒,试剂盒包括:
a)溶解在非水性溶剂中的配体,配体能够结合放射性核素并且其中溶剂选自己烷至丙三醇的相对极性范围;
b)还原剂;
c)缓冲溶液;
d)和任选地添加剂比如弱螯合剂、抗氧化剂、增溶剂或膨松剂并且其中组分a)、b),c)和d)各自为冻干形式。
在本发明优选的实施方式中,还原剂是SnCl2或SnF2或酒石酸亚锡、盐酸和水的混合物,并且缓冲溶液是磷酸盐或柠檬酸或乙酸盐缓冲溶液。可选地,缓冲剂是磷酸盐、柠檬酸或乙酸盐缓冲溶液的任何一种的组合。
优选地,弱螯合剂选自DTPA、葡庚糖酸盐、酒石酸盐和亚甲基二膦酸盐(medronate)或任何组合。抗氧化剂选自龙胆酸、抗坏血酸和对氨基苯甲酸或其组合。增溶剂选自明胶或环糊精或其组合,并且膨松剂选自甘露醇、肌醇、葡萄糖和乳糖或其组合。
组分a)、b)、c)和d)可包含在一个小瓶中。可选地,组分b)、c)和d)包含在第一小瓶中并且组分a)包含在第二小瓶中。
配体可选自ECD、HMPAO、MAG3和MIBI或其碱金属盐或其碱土金属。优选地,配体是ECDG或其碱金属盐。溶剂选自:甲醇、乙醇、乙酸乙酯、己烷、氯仿、二氯甲烷、甲苯、醚、四氢呋喃和乙腈或其组合。优选地,溶剂选自甲醇或乙醇。更优选地,溶剂是甲醇。
金属放射性核素可选自99mTc、188Re、186Re、153Sm、166Ho、90Sr、90Y、89Sr、67Ga、68Ga、111In、153Gd、59Fe,52Fe、225Ac、212Bi、45Ti、60Cu、61Cu、62Cu、64Cu、67Cu、195mPt、191mPt、193mPt、117mSn、103Pd、103mRh、89Zr、177Lu、169Er、44Sc、155Tb、140Nd、140Pr、198Au、103Ru、131Cs、223Ra、224Ra和62Zn。
优选地,放射性核素是99mTc、103Pd、103mRh、195mPt、193mPt、191Pt。更优选地,放射性核素是99mTc。
试剂盒进一步包括使用说明书。
附图说明
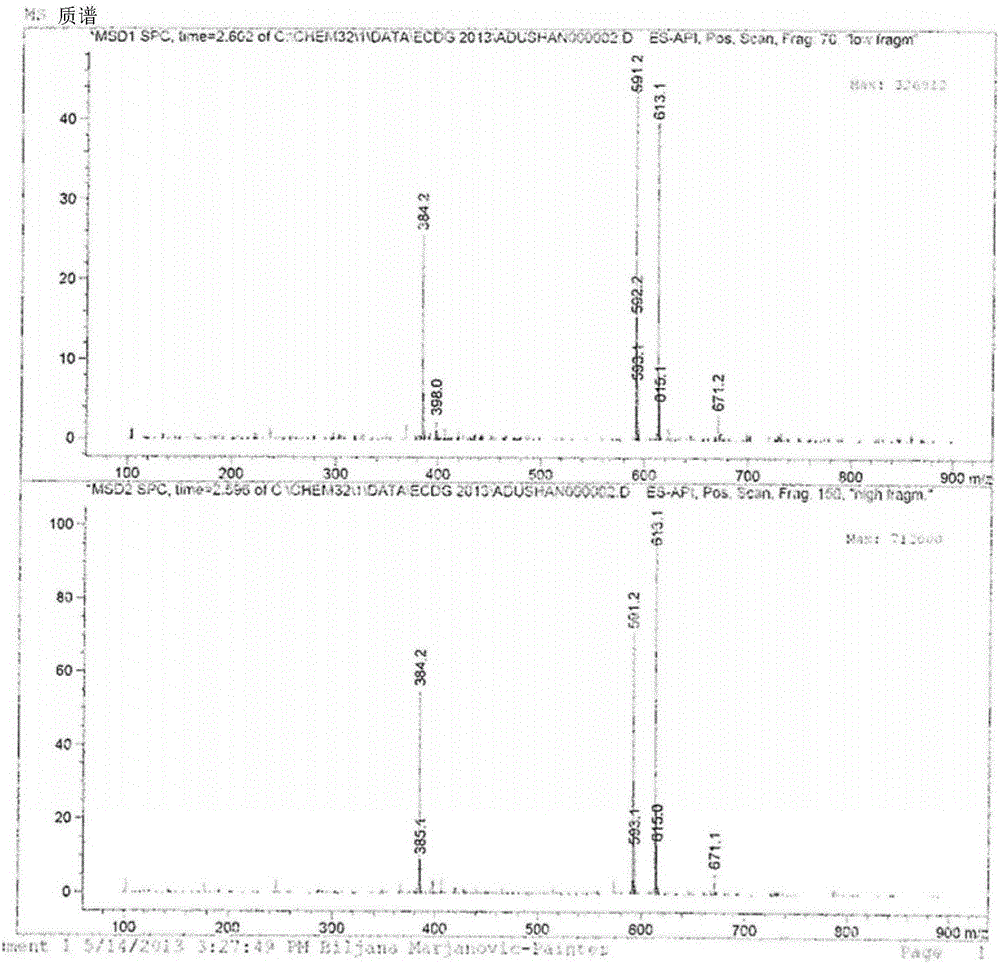
图1是ECDG产生的质谱。
具体实施方式
根据下述制备试剂盒。
在Ar(g)条件下制备下述溶液以确保没有CO2或O2:
a)足够量的ECDG或其盐溶于己烷至甘油的相对极性范围内的非水性溶剂。
b)在适当的pH下用于最佳标记条件的磷酸盐/柠檬酸缓冲溶液。
c)在中性或酸性介质中的亚锡盐溶液,所述介质用作VII氧化态的高锝酸盐离子(99mTcO4-)至IV的还原剂,以确保99mTc是化学活性的,以与配体,ECDG结合。
对于两个小瓶试剂盒配制,使用上述溶液的冷冻干燥程序涉及下述:
a)小瓶1:将足够体积的ECDG溶液添加至小瓶1,冷冻并且然后在Ar(g)条件下冷冻干燥。
b)小瓶2:将预定体积的制备的磷酸盐/柠檬酸缓冲溶液添加至充满Ar(g)的小瓶2,冷冻并冷冻干燥过夜,随后添加Sn溶液(60-100μg Sn(II)),随后在Ar(g)条件下冻干。
将所有的小瓶储存在制冷器的黑暗条件下。标记方案需要小瓶1的重构或溶解,添加小瓶1至小瓶2,随后立即添加足够的99mTc活性。将反应混合物加热(60-80℃)有限的时间,以确保标记。用TLC和HPLC的质量控制应记录>90%的标记并且放射化学纯度大于95%。
对于一个小瓶试剂盒配制,使用上述溶液的冷冻干燥程序涉及下述:
a)首先,将预定体积的制备的磷酸盐/柠檬酸缓冲剂冷冻并冷冻干燥。接着,将Sn溶液(60-100μg Sn(II))添加至充满Ar(g)的小瓶并且冷冻,随后在Ar(g)条件下冷冻干燥。
b)最后,将纯的ECDG溶于a)的冷冻干燥材料上方的非水性溶剂、冷冻并冷冻干燥。该标记方案需要仅仅通过添加足够的99mTc活性重构。将反应混合物加热(60-80℃)有限的时间,以确保标记。用TLC和HPLC的质量控制应记录>90%标记并且放射化学纯度大于95%。添加至试剂盒并且构建准备注射。
在制备试剂盒时,申请人合成制备了ECDG。从商业上可得的L-噻唑烷-4-羧酸开始,在五个合成步骤中成功进行了产生ECDG的合成路径。合成路径可简单总结如下。
从结构角度,99mTc-ECDG可视为由三个组分组成,即:(i)在其核心处的L,L-乙烯二半胱氨酸(EC)配体,(ii)两个靶向癌症的D-葡糖胺基团和(iii)99mTc放射性核素。EC可获得自商业上可得的L-4-噻唑烷羧酸的自由基促进的二聚化反应[10]。EC的硫醇和仲胺官能团是活性位点并已经显示分别被苄基(Bn)[11]和氯甲酸苄酯(Cbz)保护基团有效和高效保护(mask)。两个葡糖胺基团理论上可通过采用试剂氯甲酸乙酯经混合酸酐偶联反应与EC的酸部分结合。接着,可通过钠/氨溶液中偶联反应产物的全部脱保护(global deprotection)产生ECDG[8]。该反应可用苯基乙酸铵淬灭,其产生2-丙醇可溶性苯基乙酸钠盐,这使得从反应副产物适当纯化ECDG。该合成的ECDG可接着用99mTc标记并且根据需要使用。
如文献规定[10],由L-噻唑烷-4-羧酸精确进行EC 4的合成并且产生期望的产物,产率是38%。
一旦反应结束,使氨快速蒸发(沸点是-33℃)并且将所得残渣溶于水,以产生高碱性的(pH=12.0)溶液。因此,添加5M HCl,以使碱化的EC配体质子化并且使分子作为其二盐酸化合物盐沉淀,这在pH 3.0–2.0内实现。将开始材料L-噻唑烷-4-羧酸溶于酸性介质中并且保留在溶液中并且所以该步骤用作EC 4纯化的第一阶段。接着,将沉淀EC 4过滤并且发现该粗EC 4立即从沸腾乙醇中重结晶,随后在高真空度下干燥材料,产生纯的EC 4,为粉末状白色固体。在D2O中进行EC 4的NMR,必要时添加6.0当量的K2CO3,以(i)中和二盐酸化合物盐和(ii)使硫醇和酸官能团(functionality)去质子化,这使得EC4溶解并被分析。EC 4的质子和碳NMR数据与文献数据精确一致,以及确定的熔点。该数据也说明EC 4的纯度大于99%。
根据参考文献信息[10],EC 4被苄化(benzylate)并且没有观察到来自此的偏离(deviation)。该保护步骤是必要的,因为硫醇基也在计划的葡糖胺偶联反应中反应并且因此需要被保护。但是关于EC-Bn5的质子
并且因此,必须确定分析的溶剂系统和方法。实验发现EC-Bn 5完全溶于连同添加4.0当量的K2CO3的以6:4v/v比例的D2O和氘化的DMF的混合物中,K2CO3用于中和EC-Bn 5的二盐酸化合物盐并且使两个酸部分去质子化。这使得产生EC-Bn5的NMR数据并且用作首次报道的有关该化合物的质子和碳NMR图谱。质子图谱与亲本EC 4化合物的非常相似,但是包含苄基CH2质子作为4.69ppm处的单峰并且在7.16ppm处出现的十个芳族质子作为多峰。碳NMR图谱与质子NMR图谱的发现相关,因为在35.9ppm处观察到CH2碳原子并且在127.1ppm、128.6ppm、128.8ppm和138.6的信号源自芳族环。该数据,以及匹配在预期文献范围内的确定的熔点,确认了成功实现了苄基保护。
用氯甲酸苄酯保护基团保护EC-Bn 5的仲胺部分。与硫醇基团类似,这些仲氨基也在计划的葡糖胺偶联反应中反应并且所以也需要封端(cap)。EC-Bn Cbz保护起初在0℃下进行2h并且然后在室温(RT)下持续16h。需要二乙醚洗涤步骤,以去除任何未反应的CbzCl,随后使水性介质酸化至pH 3.0,以使EC-Bn-Cbz 6的羧酸基团质子化,
其产生产物的沉淀,为白色固体。发现,产物溶于大量的有机溶剂并且用乙酸乙酯提取酸化溶液使得EC-Bn-Cbz6被分离。随后有机相的分离和溶剂去除产生期望产物,为无定形固体。EC-Bn-Cbz 6必须在存在高真空度的情况下充分干燥,以确保材料完全没有痕量的溶剂或水。EC-Bn-Cbz 6产物在二氧化硅凝胶上快速分解并且因此可不用另外纯化,发现这与公开的数据[12]相反。用于EC-Bn-Cbz 6的NMR分析的溶剂系统可能不是确定的并且这也与在CDCl3中产生NMR数据的文献信息不一致。该产物的LC-MS分析也证明是不成功的,原因是用于MS测定的非常有问题的苄基保护基团。因此粗EC-Bn-Cbz 6直接用于接下来的步骤。
采用氯甲酸乙酯作为偶联剂进行EC-Bn-Cbz 6和四-乙酰葡糖胺的偶联反应。反应条件和流程以与现有技术中发现的那些相同的方式进行,但是确定了新的柱纯化溶剂系统。发现以(1-5):(10-90):(10-80)范围内的比例的甲醇(MeOH)、乙酸乙酯(EtOAc)和己烷的三组分溶剂组合使得完全保护的ECDG 7以较高的纯度被分离。
最后的步骤是充分保护的ECDG7的钠/氨促进的完全脱保护,以产生ECDG 3。充分保护的ECDG 7与20.0当量的钠金属反应,以完全去除酸酯、Cbz和Bn保护基团。接着,添加12.0当量的苯基乙酸铵——其使得形成苯基乙酸钠作为副产物——淬灭反应。一旦在氩气气氛蒸发了氨液体,通过2-丙醇洗涤步骤,从反应混合物去除苯基乙酸钠。苯基乙酸钠高度溶于2-丙醇,但是ECDG 3不是,并且因此在惰性气氛下过滤有机介质,以产生ECDG 3,为乳白色、强烈气味的固体。接着,用二乙醚洗涤该ECDG 3,并且然后在高真空度下避光干燥1h。通过MS确定该ECDG的鉴定和纯度(图1)并且在591.1单位观察到ECDG 3必要的MS峰。在氩下,避光-20℃储存ECDG 3。
实施例
实施例1–双小瓶试剂盒
冻干方案
a)向磷酸氢二钠(sodium phosphate dibasic)(0.284g,0.002mol)的水溶液(脱氧的(de-oxynated))添加柠檬酸(0.201g,0.001mol),以产生pH 5.5de磷酸盐/柠檬酸缓冲溶液。将855μl制备的磷酸盐/柠檬酸缓冲溶液添加至第一填充氩的小瓶,封闭并且冷冻,然后冷冻干燥过夜。
b)将盐酸(0.10ml,0.1M)添加至氯化锡(II)二水合物溶液(0.01g,0.04mmol),并且用水(脱氧的)稀释至10ml。接着,将100μl Sn溶液(=60μg Sn(II))添加至小瓶1,冷冻,随后冷冻干燥。
c)将甲醇(1.5ml)添加至第二个填充氩的具有ECDG(10mg,0.017mmol)的小瓶。将小瓶浸入液氮,以冷冻溶剂并冷冻干燥。小瓶应保持在黑暗和制冷器中。
注意,所以的小瓶应填充Ar,以确保没有CO2或O2。
标记方案
a)将355μl H2O添加至冷冻干燥的ECDG小瓶。
b)转移至冷冻干燥的缓冲剂/Sn小瓶并且加入小的磁力搅拌棒。形成漩涡(Vortex),以溶解缓冲盐。
c)随后立即添加500μl TcO4-(或等体积,用于约40mCi活性)。
d)放置在加热板上并且在70℃下搅拌15分钟。
e)进行TLC和HPLC-QC。
实施例2–一个小瓶试剂盒
冻干方案
a)向磷酸氢二钠(0.284g,0.002mol)的水溶液(脱氧的)添加柠檬酸(0.201g,0.001mol),以产生pH 5.5的磷酸盐/柠檬酸缓冲溶液。将855μl制备的磷酸盐/柠檬酸缓冲溶液添加至第一个填充氩的小瓶,封闭并且冷冻,然后冷冻干燥过夜。
b)将盐酸(0.10ml,0.1M)添加至氯化锡(II)二水合物溶液(0.01g,0.04mmol)并且用水(脱氧的)稀释至10ml。接着,将100μl Sn溶液(=60μg Sn(II))添加至小瓶1,冷冻随后冷冻干燥。
c)将甲醇(1.5ml)添加至第二个填充氩的具有ECDG(10mg,0.017mmol)的小瓶。这被定量转移至包含Sn/缓冲剂的小瓶1。将小瓶浸入液氮,以冷冻溶剂并冷冻干燥。小瓶应保持在黑暗和制冷器中。
注意,所有的小瓶应填充Ar,以确保没有CO2或O2。
标记方案
a)将500μl TcO4-(或等体积,用于约40mCi的活性)添加至包含ECDG、Sn和缓冲剂的小瓶。
b)放置在加热板上并且在70℃下搅拌15分钟。
c)进行TLC和HPLC-QC。
实施例3–合成ECDG
合成L,L-乙烯双半胱氨酸.2HCl
将L-噻唑烷-4-羧酸(30.0g,225mmol)缓慢添加至配备有冷却冷凝器(填充有液氮)、氩气体入口和填充油的出口捕集器(outlet trap)的双颈圆底烧瓶的液氨(150ml)中。强力搅拌混合物,直到所有L-噻唑烷-4-羧酸完全溶解,随后在15分钟内逐份添加清洁的钠金属(8.00g,349mmol,1.50当量)。一旦完成钠金属的添加,观察到深蓝色,并且在室温下搅拌该溶液20分钟。接着,以抹刀尖(spatula-tip)部分小心添加氯化铵,直到混合物变成白色,并且已淬灭所有未反应的钠金属。接着,使得氨溶剂蒸发并且将所得反应残留物溶于水(200ml)并且用浓缩的HCl将pH调整至3.0,其产生乙烯双半胱氨酸的二盐酸化合物盐的沉淀,为白色固体。通过真空过滤收集产物,从沸腾乙醇重结晶并且在高真空度下干燥,以产生14.7g(38%)乙烯双半胱氨酸.2HCl 4
M.p.:252-254℃(参考251-253℃[10]);
1H NMR(400MHz,D2O和6.0当量的K2CO3):
δH=3.27(2H,t,2x CH-COOH),2.70-3.00(8H,m,2x CH2-N和2x CH2-SH重叠),2.62(2H,m,2x NH)2。
13C NMR(400MHz,D2O和6.0当量K2CO3):
δC=177.9(COOH),65.6(CH-N),44.8(CH2-N),26.8(CH2-SH)。
合成S,S’-二苄基乙烯双半胱氨酸.2HCl 5
在室温下将乙烯双半胱氨酸.2HCl 4(2.0g,6.0mmol)溶于2M NaOH(30ml)并且添加乙醇(40ml),并且强力搅拌所得溶液20分钟。将二噁烷(20ml)中的苄基氯化物(1.48g,11.7mmol,2.0当量)逐滴添加至乙烯双半胱氨酸溶液并且然后在添加结束之后搅拌另外的30min。接着,真空去除乙醇和二噁烷,并且然后用5M HCl酸化所得水性混合物的pH至pH 3.0。这产生S,S’-二苄基乙烯双半胱氨酸5的盐酸盐沉淀,其在真空下过滤并且在高真空度下干燥,产率为85%(2.7g)。
M.p.:227-228℃(参考251-253℃);
1H NMR(400MHz,D2O/DMF(6:4v/v比)和4.0当量的K2CO3):
δH=7.16(10H,m,2x CH2-C6H5),3.68(4H,s,2x CH2-C6H5)3.14(2H,t,CH-COOH),2.44-2.85(10H,m,2x CH2-N,2x CH2-SH和2x NH重叠);
13C NMR(400MHz,D2O/DMF(6:4v/v比)和4.0当量的K2CO3):
δC=179.5(COOH),138.6(Ar-C),128.0(Ar-C),128.6(Ar-C),127.1(Ar-C),62.7(CH-N),46.6(CH2-N),35.9(CH2-C6H5),34.4(CH2-SH)。
合成充分保护的乙烯双半胱氨酸脱氧葡糖胺7
将S,S’-二苄基乙烯双半胱氨酸5(6.0g,11.5mmol)溶于10%K2CO3溶液(150ml)并且在冰浴中冷却至0℃。接着,将二噁烷(150ml)中的氯甲酸苄酯的混合物快速添加至溶液,其接着在0℃下搅拌2小时。接着,去除冷却浴并且将混合物在室温下搅拌16h,然后用二乙醚(2×50ml)萃取。接着,用1M HCl小心酸化水性层至pH 3.0,其产生白色化合物沉淀。添加乙酸乙酯(200ml)并且强烈搅拌将沉淀的固体溶于该有机层。分开有机层,用无水硫酸镁干燥,过滤并且在旋转式汽化器上去除溶剂。接着,在高真空度下干燥所得透明残留物,以产生5.75g(70%产率)粗N,N’-二苄氧基羰基-S,S’-二苄基乙烯双半胱氨酸6,为无定形的固体。该化合物是对于纯化不稳定并且不溶于测试的NMR溶剂,并且因而直接用于接下来的反应。
将EC-Bn-CBz6(1.34g,1.87mmol)溶于具有三乙胺(0.378g,3.74mmol,2.0当量)的干燥氯仿(30ml),并且在氩气氛下在氯化钠/冰浆冷却浴(ice slurry cooling bath)中将溶液冷却至-15℃。逐滴添加氯甲酸乙酯(0.406g,3.74mmol,2.0当量)并且搅拌所得混合物另外的15分钟。向该反应混合物,添加四乙酰葡糖胺(1.58g,4.11mmol,2.2当量)和三乙胺(0.416g,4.11mmol,2.0当量)的干燥氯仿(30ml)的溶液,并且在0℃下搅拌组合的反应混合物1h,并且然后在室温下搅拌12h。接着,将用1M HCl(2×25ml)、5%K2CO3溶液(2×25ml)、H2O(50ml)洗涤溶液,用无水硫酸镁干燥,过滤并且真空去除溶剂。通过柱色谱(二氧化硅凝胶60;流动相:MeOH/EtOAc/己烷)纯化残留物,以产生1.80(70%产率)g的完全保护的乙烯双半胱氨酸脱氧葡糖胺7,为白色结晶固体。
1H NMR(400MHz,CDCl3):
δH=8.62(2H,s,2x NH),7.48-7.40(20H,m,2x OCH2-C6H5,2x SCH2-C6H5),6.04(2H,d,四氢吡喃端基质子),5.45-5.20(6H,m,2x OCH2-C6H5,2x四氢吡喃质子重叠),4.48-4.07(6H,m,2x CH-CONH,4x四氢吡喃质子重叠),3.72-3.48(12H,4x四氢吡喃质子,4x CH2-N-,2x CH2-S-重叠),2.20-1.92(24H,8x OCH3)。
合成乙烯双半胱氨酸脱氧葡糖胺3
在氩气氛下将充分保护的乙烯双半胱氨酸脱氧葡糖胺7(1.00g,0.73mmol)溶解在氨液体(100ml)中,并且以小部分添加清洁的钠金属(0.334g,14.5mmol,20.0当量)。反应混合物变成深蓝色并且在室温下搅拌15分钟,然后添加少量的铵苯基乙酸酯,以淬灭未反应的钠金属。在氩气体流下干燥所得乳白色溶液,以提供强烈气味的乳白色固体的。在惰性气氛下避光处理粗产物。将2-丙醇(200ml)添加至材料并且强力搅拌10min,然后真空过滤。用二乙醚洗涤所得乳白色沉淀,并且然后在高真空度下干燥2小时,以产生0.230g(53%产率)的乙烯双半胱氨酸脱氧葡糖胺的钠盐(ECDG)3。通过1H NMR、HPLC和MS分析确认产物,其与文献数据一致。C20H38O12N4S2需要590.665,其中观察到591.1。
参考文献
1.http://clinicaltrials.gov/show/NCT01394679,20/08/2013
2.Zhang,Y.H.,Bryant,J.,Kong,F.L.,Yu,D.F.,Mendez,R.,Kim,E.E.&Yang,D.J.,2012,Molecular Imaging of Mesothelioma with 99mTc-ECG and 68Ga-ECG,Journal of Biomedicine and Biotechnology,2012.
3.Zaman,M.,2007,99mTc-EC-deoxyglucose–a poor man’s 18F-FDG:what will be the future of PET in molecular imaging?,European Journal of Nuclear Medicine and Molecular Imaging,34,427-428.
4.http://www.health24.com/Medical/Cancer/Facts-and-figures/South-Africa-78-increase-in-cancer-by-2030-20120721,20/08/2013.
5.Yang,D.,Kim,C.,Schechter,N.R.,Azhdarinia,A.,Yu,D.,Oh,C.,Bryant,J.L.,Won,J.,Kim,E.&Podoloff,D.A.,2003,Imaging with 99mTc-ECDG targeted at the multifunctional glucose transport system:feasibility study with rodents,Radiology,226,465-473.
6.Zhang,Y.,Mendez,R.,Kong,F.,Bryant,J.,Yu,D.,Kohanim,S.,Yang,D.&Kim,E.,2011,Efficient synthesis of 99mTc-ECDG for evaluation of mesothelioma,Journal of Nuclear Medicine,52(Suppl.1),1532
7.Ebrahimabadi,H.,Lakouraj,M.M.,Johari,F.,Charkhlooie,G.A.,Sadeghzadeh,M.,2006,Synthesis,characterization and biodistribution of99mTc-(EC-DG),a potential diagnostic agent for imaging of brain tumors,Iranian Journal of Nuclear Medicine,14(Suppl.1),36-37
8.Yang,D.J.,2008,US Patent Application US20080107598
9.Blondeau,P.,Berse,C.,Gravel,D.,1967,Dimerization of an intermediate during the sodium in liquid ammonia reduction of L-thizolidine-4-carboxylic acid,Canadian Journal of Chemistry,45,49-52
10.Assad,T.,2011,Synthesis and Characterization on novel benzovesamicol analogues,Turkish Journal of Chemistry,35,189-200.
11.Mang’era,K.O&Verbruggen,A.,1999,Synthesis and Evaluation ofβ-Homocysteine Derivatives of 99mTc-L,L-EC and 99mTc-L,L-ECD,Journal of Labelled Compounds and Radiopharmaceuticals,42,683-699.
- 还没有人留言评论。精彩留言会获得点赞!