用于使自由基聚合性烯属不饱和单体乳液聚合的方法与流程
聚合物分散体公知作为制造涂料组合物,例如石膏,底灰(renders),粘合剂,油漆,地毯背涂层,纸张涂层,工程织物的涂层等的粘结剂,并且被大量商业制造。使用水性油漆体系的优势包括成本低,容易应用和清洁,干燥时间减少,和气味或挥发性有机化合物(VOC)的排放物少或没有。
在大多数情况下,这种体系借助于通常为连续、间歇或半间歇法形式的乳液聚合制备。间歇法表示不连续的按次加料方式的聚合反应,而半间歇法表示间歇和连续方式的组合,其中在聚合反应过程中缓慢添加一部分单体。
聚合物分散体制造中的重要方面为制造效率,出于商业原因,有利的是聚合物分散体的制造时间尽可能的短。
但是一个特别的问题是去除放热聚合期间的反应热,但是该多相混合物通常包含约30%至约60%的水。热的去除经常是为提高时空产率而更迅速进行聚合的限制因素。
另外的要求是所得聚合物分散体是可高度再现的,并且就应用性能而论显示良好性能,例如由特别限定的平均分子量、分子量分布或粘度表示的。在自由基乳液聚合的情况下,运行可以由许多工艺参数控制,所述工艺参数例如聚合温度,反应物计量速率,反应热去除,反应体系的组分的性质和量,或聚合反应器设计。
通常,高聚合温度能够使聚合时间更短,但是可能产生性能劣化的产物,而较低的聚合温度可能需要更长的聚合时间,但是可能产生性能提高的产物。因此,通过升高聚合温度,通常难以减少聚合时间,同时保持所得聚合物的良好应用性能。现有技术描述了解决自由基聚合中的上述问题的不同方法。
一种方法是基于使用额外的内部冷却器或热交换器来去除反应热。例如,JP-A-07/082304描述氯乙烯在装有回流冷凝器的反应器中的悬浮聚合。为缩短聚合时间,在达到60%的总转化率之前向反应混合物中添加高活性油溶性引发剂。
WO-A-2005/016977公开一种用于通过自由基引发乳液聚合制备水性聚合物分散体的方法。该方法包括至少一种烯属不饱和化合物在至少一种分散剂和至少一种水溶性和一种油溶性自由基引发剂存在下的单步聚合。根据WO-A-2005/016977,组合使用水溶性和油溶性自由基引发剂允许制造具有低残留单体含量的水性聚合物分散体,因此避免了单独后聚合处理的必要性。
EP 2 088 162 A1公开一种用于使自由基聚合性烯属不饱和单体在较高温下,即100℃至160℃的乳液聚合的方法,其中乳液聚合在乳化剂存在下进行。
但是,上述方法通常具有聚合反应在经常损害产品性能的较高反应温度下进行的缺点,例如聚合物情况下分子量降低,油漆情况下抗湿擦性降低,或粘合剂情况下耐热性降低。与已经在较低温度下制备的产物相比,这里主要问题总是除热的手段必须是商业上可行的,同时不损害产品性能。
因此,本发明的技术问题为提供改善的乳液聚合方法,其在低温下比常规乳液聚合方法更快速,以及在相同时间下,产生具有所需分子量和具有良好性能(就应用性能而论)的产物。另外,本发明的另一个技术问题为提供改善的乳液聚合方法,其产生就应用性能而论具有优越性能,同时需要可与常规乳液聚合方法相当的聚合时间的产物。
根据本发明,通过提供一种用于使自由基聚合性烯属不饱和主要单体和任选可与之共聚的其它辅助单体乳液聚合的方法解决了这些技术问题,其中该聚合包括这样的聚合反应阶段,其始于第一次添加引发剂或始于第一次添加单体,以后到者为准,并且止于添加完引发剂或添加完全部单体,以后到者为准,其中反应温度在所述聚合反应阶段期间从约30℃至约85℃的起始温度升高到约60℃至约160℃的结束温度,和其中在所述聚合反应阶段期间的至少25%,聚合温度升高。
温度曲线
根据本发明,单体由乳液聚合方法聚合,其中反应温度在特别地定义的聚合反应阶段期间从约30℃至约85℃的起始温度升高到约60℃至约160℃的结束温度。
意外地发现通过使用本发明的方法,可以更迅速地进行聚合,由此提高时空产率,同时在相同时间下,获得具有所需分子量和具有良好性能(就应用性能而论)的产物。另外,可以获得就应用性能而论具有优越性能,同时需要与常规乳液聚合方法相当的聚合时间的产物。根据本发明,这一点通过在较低聚合温度下引发,然后在聚合反应过程中升高聚合温度来实现。因此,与常规的乳液聚合的低温方法相比,本发明方法的特征为聚合时间显著减少,或当应用常规聚合时间时应用性能提高。
在本发明的涵义内,术语“聚合反应阶段”表示始于第一次添加引发剂或第一次添加单体(以后到者为准)的阶段。因此,仅当存在单体和引发剂两种组分时,聚合反应阶段才开始。合适的自由基聚合反应引发剂包括例如热引发剂,光化学引发剂和氧化还原引发剂体系。当使用氧化还原引发剂体系时,第一次添加引发剂表示添加氧化还原引发剂体系(氧化剂和/或还原剂)的第二组分,因为仅在两种组分存在时才形成自由基。应注意上述“聚合反应阶段”的起点并不必然与取决于引发自由基形成的聚合反应的实际起点一致。例如,当使用热引发剂时,在达到热引发剂的分解温度之前,即使存在引发剂和单体,但聚合反应并未开始。聚合反应的实际起点可以例如通过鉴别反应混合物中的自由基(例如通过ESR光谱学)或通过聚合反应热的出现来加以确定。
通常对于大多数乳液聚合方法(特别是对于商业方法)而言,有利的是“聚合反应阶段”的起点(如以上定义)和聚合反应的实际起点之间的时间间隔尽可能的短。
上述聚合反应阶段止于添加完引发剂或添加完全部单体(即主要单体和辅助单体),以后到者为准。
要求保护的乳液聚合方法可以包括聚合反应阶段之后的任选后加热,以完成聚合反应。根据本发明的优选实施方案,后加热在至少85℃下进行至少15min。根据特别优选实施方案,后加热在至少90℃的温度下进行约15至约60min。例如在任选后加热之后,在基于总重量直至98%的单体,优选多至99%的单体,特别优选多至99.5%的单体聚合之后,要求保护的乳液聚合方法通常终止。换言之,当基于全部单体,多至98wt%,优选多至99wt%,特别优选多至99.5wt%的单体已经聚合时,要求保护的乳液聚合方法结束或被认为结束。
因此,要求保护的乳液聚合方法不同于去除残余单体的任选后聚合反应(优选在不同反应器中进行),即使对于这种后聚合可以添加额外的引发剂。本领域技术人员可以清楚地区别实际聚合反应与这种任选的后聚合反应。基于全部单体,残余单体通常不超过约2wt%,优选不超过约1wt%,特别优选不超过约0.5wt%。因此,后聚合反应可以任选接着要求保护的乳液聚合方法之后进行,其中在基于全部单体,多至98wt%,优选多至99wt%,特别优选多至99.5wt%的单体已经聚合之后,后聚合反应开始。
根据本发明,聚合反应阶段的持续时间优选为至多5小时,更优选少于3.5小时,和甚至更优选少于2小时。
此外,根据本发明,术语“起始温度”表示聚合反应阶段开始时的温度。“结束温度”定义为聚合反应阶段结束时的温度。
起始温度为约30℃至约85℃。在本发明的优选实施方案中,起始温度为约40℃至约80℃。在本发明的特别优选实施方案中,起始温度为约55℃至约70℃。结束温度为约60℃至约160℃,优选为约80℃至约120℃。在特别优选的实施方案中,结束温度为约85℃至约110℃,在另一个特别优选实施方案中,结束温度为约105℃至约120℃。
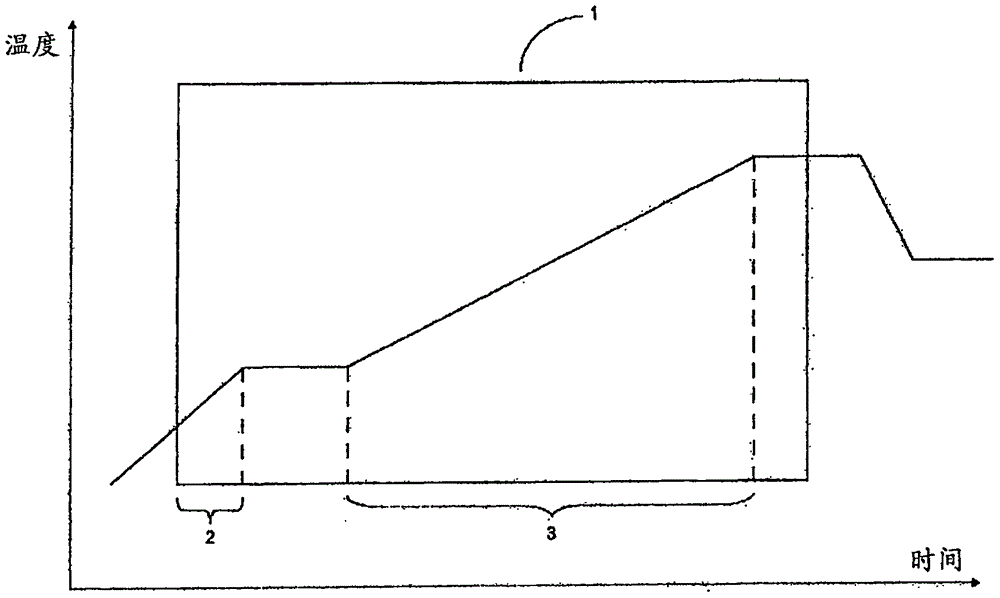
本发明的聚合反应由图1中所示的温度曲线举例说明。该附图显示矩形(1)形式的聚合反应的聚合反应阶段。聚合反应阶段的起点由第一次添加引发剂或第一次添加单体定义,以后到者为准。聚合反应阶段的终点由添加完引发剂或添加完单体定义,以后到者为准。图1中所示聚合反应阶段的实例包含具有第一次升温的时期(2)和具有第二次升温的时期(3)。
反应温度的升高可以按照任何合适的温度曲线,其中结束温度高于起始温度。优选,温度连续或半连续升高。在特别优选的实施方案中,温度连续升高。
术语“连续升高”表示温度随时间连续(以数学含义)升高,即图形为单一不间断曲线,没有“缺口”或“跳跃”。图2中示出反应温度的连续升高实例,包括
(a)反应温度具有恒定斜率的线性升高,
(b)反应温度具有两个或多个不同斜率的线性升高,和
(c)反应温度的非线性升高。
反应温度的半连续升高包括所有合适的温度曲线,其中温度随时间升高,但是其中至少2个伴随温度升高的聚合反应时期被没有温度升高的聚合反应时期间隔。
反应温度的半连续升高的实例在图3中示出,包括具有至少2个斜率相同或不同的线性或非线性升温的反应时期,其中该至少2个反应时期被恒温反应时期间隔(参见图3a和3b)。因此,半连续升高还包含反应温度的逐步升高,包括例如至少2步升温,至少3步升温或至少4步升温或至少5步升温。根据特别优选实施方案,半连续升高包含反应温度的逐步升高,包括至少10步升温或至少15步升温。图3c示例性地显示这种包括4步升温的逐步升温。根据本发明,包括至少20步升温的反应温度的逐步升高定义为连续升温。
升温期间的温度速率优选为约0.01℃/min至约10℃/min。在更优选实施方案中,温度速率为约0.05℃/min至约5℃/min。在特别优选实施方案中,温度速率为约0.2℃/min至约3℃/min。应注意温度速率无须恒定。聚合反应的不同阶段也可以具有不同的温度速率。在本发明的特别优选的实施方案中,升温包括温度速率大于0.05℃/min并且小于0.2℃/min的时期,以及温度速率为0.2℃/min至0.7℃/min的后续时期,和其中在所述聚合反应阶段过程中在升温期间添加基于总重量至少75%的全部单体。随着聚合反应阶段开始,反应温度可以已经升高。替代地,反应温度可以在聚合反应阶段开始之后最初保持恒定,然后例如在聚合反应阶段持续时间的第一个75%,优选第一个50%,特别优选第一个30%内开始升温。
升温的持续时间
在至少25%的聚合反应阶段期间,聚合温度升高。这意味着在至少25%的所述聚合反应阶段持续时间期间,聚合温度在所述聚合反应阶段期间升高。根据本发明的优选实施方案,在至少50%,更优选至少60%的聚合反应阶段持续时间,聚合温度升高。根据本发明的特别优选实施方案,在至少70%的聚合反应阶段持续时间,聚合温度升高。
当回到图1时,可见所示聚合反应阶段(1)包含2个升温时期(2)和(3)。根据本发明,这2个时期(2)和(3)的持续时间总和占聚合反应阶段(1)的持续时间的至少25%。
在连续升高的情况下,升温的持续时间在图2中用(a)表示。在反应温度半连续升高的情况下,反应温度的升高持续时间仅表示其中温度升高的那些反应时期的持续时间总和(在图3中用(b)表示)。
借助于这一必要的特征,可以获得本发明方法的有益效果,即减少聚合时间,而同时获得具有良好性能(就应用性能而论)的产物。另外,当聚合时间保持恒定时,也可获得性能改善的产物。
单体的添加
根据本发明的优选实施方案,在所述聚合反应阶段过程中在升温期间添加基于总重量至少25%的全部单体(即主要单体和辅助单体)。根据更优选实施方案,在所述聚合反应阶段过程中在升温期间添加基于总重量至少40%,更优选至少65%的全部单体。在特别优选的实施方案中,在所述聚合反应阶段过程中在升温期间添加基于总重量至少75%的全部单体。
假定聚合反应在饥饿条件下进行,在聚合反应阶段过程中添加的单体基本立刻反应,并转化成各个(共)聚合物。因此,根据优选实施方案,基于总重量,至少25%,更优选至少40%,更优选至少65%,和特别优选至少75%的全部单体在升温过程中聚合/转化。因此,单体的转化率可以由聚合反应过程中添加的单体量计算。可选地,可以例如通过以适当时间间隔从反应混合物中取样并借助于已知方法分析该试样来确定总转化率。
单体
本发明涉及一种用于使自由基聚合性烯属不饱和主要单体和任选的可与之共聚的其它辅助单体乳液聚合的方法。当聚合仅一种单体时,由本发明的方法获得均聚物。当聚合至少2种单体时,由本发明的方法获得共聚物。在要求保护的方法的优选实施方案中,至少2种单体聚合获得共聚物。
主要单体
主要单体通常形成首要单体,并优选占待聚合的全部单体的至少50wt%,更优选至少约75wt%。
自由基聚合性烯属不饱和主要单体包含可通过自由基聚合进行聚合的所有可能的烯属不饱和单体。
自由基聚合性烯属不饱和单体优选选自C1-C18链烷酸的乙烯基酯,芳香族酸的乙烯基酯,乙烯基芳香族化合物,乙烯基卤化物,α-烯烃,二烯,烯属不饱和单羧酸或二羧酸与烷醇的单酯和二酯,及其组合。根据本发明的实施方案,将苯乙烯和1,3-丁二烯的组合排除出上述自由基聚合性烯属不饱和单体的可能组合。
合适的乙烯基酯包括例如具有1至18个碳原子的直链或支化羧酸的乙烯基酯,芳香族酸的乙烯基酯,例如甲酸乙烯酯,乙酸乙烯酯,丙酸乙烯酯,丁酸乙烯酯,异丁酸乙烯酯,新戊酸乙烯酯,乙酸1-甲乙烯酯,2-乙基己酸乙烯酯,月桂酸乙烯酯,棕榈酸乙烯酯,肉豆蔻酸乙烯酯,硬脂酸乙烯酯,具有5至11个碳原子的α-支化羧酸的乙烯基酯,特别是具有9至11个碳原子的叔碳酸(Versatic acid)的乙烯基酯(即VeoVa9,VeoVa10,VeoVa11),苯甲酸乙烯酯,苯甲酸4-叔丁基乙烯酯,及其组合。乙酸乙烯酯,月桂酸乙烯酯和具有9至11个碳原子的叔碳酸的乙烯基酯(即VeoVa9,VeoVa10,VeoVa11)是特别优选的。
合适的乙烯基芳香族化合物单体的实例包括苯乙烯,乙烯基甲苯,和α-甲基苯乙烯。苯乙烯是特别优选的。
合适的乙烯基卤化物包括氟乙烯,偏二氟乙烯,氯乙烯,偏氯乙烯和溴乙烯。氯乙烯是特别优选的。
合适的α-烯烃或二烯单体优选具有2至6个碳原子,可以包括乙烯,丙烯,异丙烯,正丁烯,正戊烯,1,3-丁二烯和异戊二烯。乙烯是特别优选的。
合适的烯属不饱和单羧酸与烷醇的酯包括例如丙烯酸、甲基丙烯酸和巴豆酸的酯。优选的烯属不饱和单羧酸的酯包括(甲基)丙烯酸烷基酯(即丙烯酸的烷基酯或甲基丙烯酸的烷基酯)。这些的实例为丙烯酸甲酯,丙烯酸乙酯,丙烯酸正丙酯,丙烯酸异丙酯,丙烯酸正丁酯,丙烯酸叔丁酯,丙烯酸异丁酯,丙烯酸2-乙基己酯,丙烯酸环己酯,丙烯酸降冰片基酯,甲基丙烯酸甲酯,甲基丙烯酸乙酯,甲基丙烯酸正丙酯,甲基丙烯酸异丙酯,甲基丙烯酸正丁酯,甲基丙烯酸叔丁酯,甲基丙烯酸异丁酯,甲基丙烯酸2-乙基己酯,甲基丙烯酸环己酯,和甲基丙烯酸降冰片基酯。这些酯可以单独使用或以两种或多种酯的组合形式使用。丙烯酸甲酯,甲基丙烯酸甲酯和丙烯酸2-乙基己酯是特别优选的。
合适的烯属不饱和二羧酸与烷醇的单酯和二酯的实例包括富马酸、马来酸和衣康酸的单酯和二酯,例如二乙酯和二异丙酯,特别是马来酸二丁酯,马来酸二辛酯,马来酸单辛酯,和马来酸酐。
优选的共聚物
上述单体可以单独使用或组合使用,以形成各种均聚或共聚物。优选的均聚物为乙酸乙烯酯聚合物和甲基丙烯酸甲酯和丙烯酸酯聚合物。但是,优选使用单体的组合来形成以下共聚物之一:
(1)根据本发明的优选实施方案,形成乙烯基酯-乙烯共聚物,例如乙酸乙烯酯-乙烯共聚物,其任选包含一种或多种选自氯乙烯,其它乙烯基酯,烯属不饱和二羧酸与烷醇的二酯,及其组合的其它单体。其它乙烯基酯优选选自丙酸乙烯基酯,月桂酸乙烯基酯,新戊酸乙烯基酯,2-乙基己酸乙烯基酯,和具有5至11个碳原子的α-支化羧酸的乙烯基酯,特别是具有9至11个碳原子的叔碳酸的乙烯基酯(即VeoVa9,VeoVa10,VeoVa11),及其组合。烯属不饱和二羧酸与烷醇的二酯优选选自马来酸二丁酯或马来酸二辛酯。
在特别优选的实施方案中,形成乙酸乙烯酯和乙烯的共聚物,其中该共聚物包含1wt%至40wt%,更优选5wt%至28wt%的乙烯,基于单体总量。当另外使用氯乙烯,其它乙烯基酯,或烯属不饱和二羧酸与烷醇的二酯时,可以以基于单体总量,1至60wt%的量包含这些组分。例如,共聚物可以为乙酸乙烯酯,基于单体总量,1wt%至40wt%的乙烯,1wt%至60wt%的氯乙烯的共聚物。根据另一个实例,共聚物可以为乙酸乙烯酯,基于单体总量,1wt%至40wt%的乙烯,1wt%至60wt%的具有9至11个碳原子的叔碳酸的乙烯基酯的共聚物。
(2)根据本发明的另一个优选实施方案,形成乙酸乙烯酯和至少一种其它单体的共聚物,所述其它单体选自乙烯,氯乙烯,其它乙烯基酯,丙烯酸酯,甲基丙烯酸酯,烯属不饱和二羧酸与烷醇的二酯,及其组合。其它乙烯基酯优选选自丙酸乙烯基酯,月桂酸乙烯基酯,新戊酸乙烯基酯,2-乙基己酸乙烯基酯,和具有5至11个碳原子的α-支化羧酸的乙烯基酯,特别是具有9至11个碳原子的叔碳酸的乙烯基酯(即VeoVa9,VeoVa10,VeoVa11),及其组合。丙烯酸酯或甲基丙烯酸酯优选选自甲基丙烯酸甲酯,丙烯酸甲酯,丙烯酸丁酯,其中使用的丙烯酸丁酯可以为丙烯酸正、异或叔丁酯,以及丙烯酸2-乙基己酯,及其组合。烯属不饱和二羧酸与烷醇的二酯优选选自马来酸二丁酯或马来酸二辛酯。优选以基于单体总量,1至40wt%的量包含至少一种其它单体。
在特别优选的实施方案中,形成基于单体总量,30wt%至75wt%的乙酸乙烯酯,1wt%至30wt%的月桂酸乙烯基酯或具有9至11个碳原子的叔碳酸的乙烯基酯,1wt%至30wt%的丙烯酸正丁酯或丙烯酸2-乙基己酯,和1wt%至40wt%的乙烯的共聚物。
(3)根据本发明的另一个实施方案,形成任选进一步包含乙烯的乙烯基酯-丙烯酸酯共聚物,或任选进一步包含乙烯的乙烯基酯-甲基丙烯酸酯共聚物。乙烯基酯可以选自乙酸乙烯酯,丙酸乙烯酯,月桂酸乙烯酯,新戊酸乙烯酯,2-乙基己酸乙烯酯,和具有5至11个碳原子的α-支化羧酸的乙烯基酯,特别是具有9至11个碳原子的叔碳酸的乙烯基酯(即VeoVa9,VeoVa10,VeoVa11),及其组合。丙烯酸酯或甲基丙烯酸酯可以选自甲基丙烯酸甲酯,丙烯酸甲酯,丙烯酸丁酯,其中使用的丙烯酸丁酯可以为丙烯酸正、异或叔丁酯,以及丙烯酸2-乙基己酯,及其组合。
例如,形成乙酸乙烯酯,1wt%至60wt%的丙烯酸正丁酯或丙烯酸2-乙基己酯,和任选1wt%至40wt%的乙烯的共聚物。
(4)根据本发明的另一个实施方案,形成任选进一步包含1,3-丁二烯或苯乙烯的丙烯酸酯或甲基丙烯酸酯共聚物。丙烯酸酯或甲基丙烯酸酯可以选自甲基丙烯酸甲酯,丙烯酸甲酯,丙烯酸丁酯,其中使用的丙烯酸丁酯可以为丙烯酸正、异或叔丁酯,以及丙烯酸2-乙基己酯,及其组合。
例如,形成甲基丙烯酸甲酯和丙烯酸2-乙基己酯的共聚物,或甲基丙烯酸甲酯和1,3-丁二烯的共聚物,或苯乙烯和丙烯酸丁酯的共聚物,或苯乙烯和甲基丙烯酸甲酯和丙烯酸丁酯的共聚物,或苯乙烯和丙烯酸2-乙基己酯的共聚物。
根据本发明,最优选的共聚物选自乙烯基酯-乙烯共聚物,例如乙酸乙烯酯-乙烯共聚物,和乙酸乙烯酯和乙烯以及具有9至11个碳原子的叔碳酸的乙烯基酯(即VeoVa9,VeoVa10,VeoVa11)的共聚物。
辅助单体
形成均聚或共聚物的上述单体仅表示主要单体。另外,合适的辅助单体可以与主要单体一起共聚合。因此,除上述主要单体之外,可以使用其它助共聚单体,其以特殊方式改性聚合物性能。这种辅助单体优选以基于待聚合的全部单体,小于20wt%,更优选约0.01wt%至约15wt%,和甚至更优选约0.1wt%至约10wt%的量共聚合。
辅助单体优选选自烯属不饱和单和二羧酸,烯属不饱和磺酸或其盐,烯属不饱和羧酸酰胺和腈,烯属不饱和膦酸或其盐,烯属不饱和亚乙基脲衍生物,烯属不饱和1,3-二羰基化合物,烯属不饱和含羟基或环氧基单体,烯属不饱和硅烷化合物,及其组合。
另外,组合物可以包含预交联单体,例如多烯属不饱和共聚单体,实例为己二酸二乙烯酯,马来酸二烯丙酯,邻苯二甲酸二烯丙酯,甲基丙烯酸烯丙酯,二丙烯酸丁二醇酯,和氰脲酸三烯丙酯,或后交联共聚单体,实例为丙烯酰胺基乙醇酸(AGA),甲基丙烯酰基酰胺基乙醇酸甲酯(MAGME),N-羟甲基丙烯酰胺(NMA),N-羟甲基甲基丙烯酰胺,N-羟甲基烯丙基氨基甲酸酯,烷基醚和酯,例如N-羟甲基丙烯酰胺、N-羟甲基甲基丙烯酰胺和N-羟甲基烯丙基氨基甲酸酯的异丁氧基醚或酯。
合适的烯属不饱和单和二羧酸的实例包括丙烯酸,甲基丙烯酸,巴豆酸,马来酸,衣康酸,柠康酸和富马酸。合适的烯属不饱和磺酸或其盐包括例如乙烯基磺酸,2-丙烯酰胺基-2-甲基丙烷磺酸(AMPS),2-丙烯酰氧基和2-甲基丙烯酰氧基乙烷磺酸,3-丙烯酰氧基-和3-甲基丙烯酰氧基丙烷磺酸和苯乙烷磺酸。烯属不饱和膦酸或其盐的实例包括乙烯基膦酸。除所述酸之外,也可使用其盐,优选其碱金属盐或其铵盐,特别是其钠盐,例如乙烯基磺酸或2-丙烯酰胺基丙烷磺酸的钠盐。特别优选使用乙烯基磺酸的钠盐。
合适的烯属不饱和羧酸酰胺和羧酸腈包括例如丙烯腈,丙烯酰基酰胺,甲基丙烯酰基酰胺,乙酰丙酮丙烯酰胺,巴豆酰胺,富马酸的单或二酰胺,马来酸的单或二酰胺,衣康酸的单或二酰胺,和柠康酸的单或二酰胺。除酰胺之外,也可使用其N-官能化衍生物,例如N-烷基或N,N-二烷基酰胺。
合适的烯属不饱和亚乙基脲衍生物包括例如咪唑啉-2-酮的N-乙烯基和N-烯丙基脲和衍生物,例如N-乙烯基和N-烯丙基咪唑啉-2-酮,N-乙烯基氧乙基咪唑啉-2-酮,N-(2-(甲基)丙烯酰基酰胺基乙基)咪唑啉-2-酮,N-(2-(甲基)丙烯酰氧基乙基)咪唑啉-2-酮和N-(2-(甲基)丙烯酰氧基乙酰胺基乙基)咪唑啉-2-酮。也可以使用脲或咪唑啉-2-酮的其它衍生物。
合适的烯属不饱和1,3-二羰基化合物包括例如含乙酰基乙酰氧基基团的单体,例如烯丙基乙酰乙酸酯,乙烯基乙酰乙酸酯,乙酰乙酰氧基乙基丙烯酸酯,乙酰乙酰氧基乙基甲基丙烯酸酯,乙酰乙酰氧基丙基甲基丙烯酸酯,2,3-二(乙酰基乙酰氧基)甲基丙烯酸丙基酯和乙酰乙酰氧基丁基甲基丙烯酸酯。
合适的烯属不饱和含羟基或环氧基单体包括例如甲基丙烯酸羟乙酯,丙烯酸缩水甘油酯,甲基丙烯酸缩水甘油酯,烯丙基缩水甘油醚,乙烯基缩水甘油醚,丙烯酸羟丙酯,丙烯酸羟丁酯,丙烯酸羟乙酯,甲基丙烯酸羟丙酯,氧化乙烯基环己烯,氧化柠檬烯,氧化月桂烯,氧化石竹烯,在芳香族部分中用缩水甘油基取代的乙烯基甲苯和苯乙烯,在芳香族部分中用缩水甘油基取代的乙烯基苯甲酸酯。优选,烯属不饱和含羟基或环氧基单体选自甲基丙烯酸羟乙酯,丙烯酸缩水甘油酯,甲基丙烯酸缩水甘油酯,烯丙基缩水甘油醚,乙烯基缩水甘油醚,丙烯酸羟丙酯,丙烯酸羟丁酯,丙烯酸羟乙酯,甲基丙烯酸羟丙酯,及其组合。
烯属不饱和硅烷化合物的实例包括通式R1SiR0-2(OR2)1-3,其中R和OR2部分的数目使得硅为四价,其中R为C1至C3烷基,C1至C3烷氧基或卤素(例如C1或Br),R1为CH2CR3(CH2)0-1或CH2CR3CO2(CH2)1-3,R2为具有1至12个碳原子,优选1至3个碳原子的非支化或支化未取代或取代烷基,或具有2至12个碳原子的酰基,如果需要,R2可以被醚基团间隔,R3为H或CH3。优选,烯属不饱和硅烷化合物选自γ-丙烯酰基-和γ-甲基丙烯酰氧基丙基三(烷氧基)硅烷,γ-甲基丙烯酰氧基甲基三(烷氧基)硅烷,γ-甲基丙烯酰氧基丙基甲基二(烷氧基)硅烷,乙烯基烷基二(烷氧基)硅烷和乙烯基三(烷氧基)硅烷,其中使用的烷氧基可以为甲氧基,乙氧基,甲氧基亚乙基,乙氧基亚乙基,甲氧基丙二醇醚或乙氧基丙二醇醚自由基。合适的烯属不饱和硅烷化合物的实例包括乙烯基三甲氧基硅烷,乙烯基三乙氧基甲硅烷,乙烯基三丙氧基硅烷,乙烯基三异丙氧基硅烷,乙烯基三-(1-甲氧基)异丙氧基硅烷,乙烯基三丁氧基硅烷,乙烯基三乙酰氧基硅烷,3-甲基丙烯酰氧基丙基三甲氧基硅烷,3-甲基丙烯酰氧基丙基甲基二甲氧基硅烷,甲基丙烯酰氧基甲基三甲氧基硅烷,3-甲基丙烯酰氧基丙基三(2-甲氧基乙氧基)硅烷,乙烯三氯硅烷,乙烯基甲基二氯甲硅烷,乙烯基三(2-甲氧基乙氧基)硅烷,三乙酰氧基乙烯基硅烷。根据本发明的优选实施方案,辅助单体包含选自乙烯基三甲氧基硅烷,乙烯基三乙氧基硅烷,乙烯基三-(1-甲氧基)异丙氧基硅烷,甲基丙烯酰氧基丙基三(2-甲氧基乙氧基)硅烷,3-甲基丙烯酰氧基丙基三甲氧基硅烷,3-甲基丙烯酰氧基丙基甲基二甲氧基硅烷,3-甲基丙烯酰氧基甲基三甲氧基硅烷,及其组合的硅烷化合物。其它合适的烯属不饱和硅烷化合物包括例如包含乙烯基官能团的硅烷低聚物,例如 RC-1或 VX-193。
根据本发明,最优选的辅助单体选自丙烯酸,甲基丙烯酸,衣康酸,乙烯基磺酸的钠盐,邻苯二甲酸二烯丙酯,乙烯基三甲氧基硅烷,乙烯基三乙氧基甲硅烷,3-甲基丙烯酰氧基丙基三甲氧基硅烷,丙烯酸缩水甘油酯和甲基丙烯酸缩水甘油酯。特别优选使用烯属不饱和硅烷单体或烯属不饱和单或二羧酸,结合烯属不饱和含环氧基单体。最优选,将基于全部单体,0.5至2.0%的甲基丙烯酸缩水甘油酯与基于全部单体,0.05至1.5%的烯属不饱和硅烷单体结合使用。另外,还优选将基于全部单体,0.5至5.0%的甲基丙烯酸缩水甘油酯与基于全部单体,0.5至2%的衣康酸结合使用。
根据本发明的优选实施方案,使乙酸乙烯酯和一种或多种其它主要单体和/或一种或多种辅助单体共聚。优选,使50至99%的乙酸乙烯酯,1至40%的乙烯,和任选至多30%的一种或多种其它主要单体和/或至多10%的一种或多种辅助单体共聚。共聚物优选由以下混合物制备,该混合物包含基于全部单体,1至40wt%的乙烯,59.9至98.9wt%的乙酸乙烯酯,0.05至5wt%的烯属不饱和含环氧基单体,0.05至5wt%的烯属不饱和硅烷化合物,和0至20wt%的一种或多种其它主要或辅助单体。烯属不饱和含环氧基团单体优选为丙烯酸缩水甘油酯或甲基丙烯酸缩水甘油酯,烯属不饱和硅烷化合物优选为3-甲基丙烯酰氧基丙基三甲氧基硅烷,乙烯基三甲氧基硅烷或乙烯基三乙氧基硅烷。根据本发明的另一个优选实施方案,使苯乙烯,丙烯酸,和任选一种或多种其它主要单体和/或一种或多种辅助单体共聚。在上述情况中,其它主要单体优选选自丙烯酸甲酯,甲基丙烯酸甲酯,丙烯酸2-乙基己酯,丙烯酸丁酯,丙酸乙烯酯,月桂酸乙烯酯,棕榈酸乙烯酯,肉豆蔻酸乙烯酯,新戊酸乙烯酯,2-乙基己酸乙烯酯,和具有5至11个碳原子的α-支化羧酸的乙烯基酯,特别是具有9至11个碳原子的叔碳酸的乙烯基酯(即VeoVa9,VeoVa10,VeoVa11),及其组合。
选择在本发明方法中使用的单体,使得能够产生具有所需最终应用所需要的性能的聚合物或共聚物。这一点可以通过以本领域技术人员已知的方式,设定形成的聚合物的玻璃化转变温度以及相应的共聚参数来实现。
通常选择主要单体和辅助单体,使得获得约-70℃至约110℃,优选约-50℃至约70℃,更优选约-30℃至约60℃的玻璃化转变温度Tg。聚合物的玻璃化转变温度Tg可以以已知的方式,通过使用10℃/min的加热速率和确定加热曲线中点的差示扫描量热法(DSC)来测定。Tg也可以预先通过Fox公式,以常规方式大致计算。
其它硅烷的添加
也可在聚合反应阶段之前、期间或之后添加额外的水解性硅化合物。根据本发明的优选实施方案,水解性硅化合物选自水解性环氧硅烷,水解性氨基硅烷,水解性巯基硅烷,具有式(R6)n-Si-(OR7)4-n的水解性烷氧基硅烷化合物,其中n为0,1,2或3,R6和R7各自独立地为直链或支化C1-C16烷基,及其组合。
合适的水解性环氧硅烷包括例如3-环氧丙氧基丙基三甲氧基硅烷(也已知为商标 GF80)和3-环氧丙氧基丙基三乙氧基甲硅烷。其它合适的水解性环氧硅烷包括例如包含环氧官能团的硅烷低聚物,例如 CoatOSil*MP200。
合适的水解性氨基硅烷包括例如3-(2-氨基乙基氨基)丙基三甲氧基硅烷,和3-(2-氨基乙基氨基)丙基甲基二甲氧基硅烷。
合适的水解性巯基硅烷包括例如通式HS-(CR42)1-3-SiR53的巯基硅烷,其中R4相同或不同并且为H或C1至C6烷基,R5相同或不同并且为C1至C6烷基或C1至C6烷氧基,至少一个基团R5为烷氧基。优选,水解性巯基硅烷选自3-巯基丙基三乙氧基硅烷,3-巯基丙基三甲氧基硅烷,和3-巯基丙基甲基二甲氧基硅烷。
合适的水解性烷氧基硅烷化合物包括例如式(R6)n-Si-(OR7)4-n的硅烷,其中n为0,1,2或3,R6和R7各自独立地为直链或支化C1-C16烷基。优选,水解性硅烷化合物选自四甲氧基硅烷,四乙氧基硅烷,甲基三甲氧基硅烷,乙基三甲氧基硅烷,丙基三甲氧基硅烷,丁基三甲氧基硅烷,戊基三甲氧基硅烷,己基三甲氧基硅烷,甲基三乙氧甲硅烷,乙基三乙氧基硅烷,丙基三乙氧基硅烷,丁基三乙氧基硅烷,戊基三乙氧基硅烷和己基三乙氧基硅烷。特别优选的水解性硅化合物为3-环氧丙氧基丙基三甲氧基硅烷,3-环氧丙氧基丙基三乙氧基硅烷,3-(2-氨基乙基氨基)丙基三甲氧基硅烷,3-巯基丙基三甲氧基硅烷,四乙氧基硅烷,甲基三乙氧基硅烷,己基三乙氧基硅烷,及其组合。
引发剂和调节剂
烯属不饱和单体在这些单体的自由基聚合的至少一种引发剂存在下发生聚合。用于引发和使聚合反应继续的自由基聚合的适合引发剂包括能够引发多相体系中的自由基水性聚合的所有已知引发剂。这些引发剂可以为水溶性引发剂(包括不仅是水溶性,而且另外也是油溶性的那些引发剂),水不溶性引发剂(即仅油溶性的那些引发剂),或其组合。根据本发明,优选使用水溶性引发剂。
根据本发明,水溶性自由基引发剂为在20℃和大气压下,在去离子水中具有至少1wt%的溶解性的引发剂。
合适的引发剂包括例如过氧化物和偶氮化合物。水溶性自由基引发剂的实例包括无机过氧化物,包括过氧化氢和过氧二硫酸,例如单或二碱金属盐,特别是过二硫酸的各个钠或钾盐,或单或二铵盐。其它实例包括有机过氧化氢,例如枯基过氧氢或叔丁基氢过氧化物(TBHP)。水溶性自由基引发剂的其它实例包括偶氮引发剂,例如2,2′-偶氮二(2-甲基戊酮脒)二氢氯化物。
聚合反应可以例如通过热引发,由上述引发剂引发。另外,上述化合物也可以在氧化还原体系内使用,其中还原剂与上述氧化剂结合使用。可以使用的还原剂包括过渡金属盐,例如铁(II)/(III)盐,羟基甲烷亚磺酸的碱金属盐,例如甲醛次硫酸钠二水合物或2-羟基-2-亚磺酸乙酸二钠盐,2-羟基-2-磺酸乙酸二钠盐的混合物,以及亚硫酸钠(Brü FF6/FF6M和Brü FF7),链长C10-C14的硫醇,丁-1-烯-3-醇,羟胺盐,二烷基二硫代氨基甲酸钠,亚硫酸钠,亚硫酸氢钠,焦亚硫酸钠,硫代硫酸钠,亚硫酸氢铵,连二亚硫酸钠,丙酮-亚硫酸氢盐加合物,二异丙基黄原素二硫化物,抗坏血酸,酒石酸,异抗坏血酸,异抗坏血酸钠,还原糖,硼酸,脲和甲酸。
用于氧化还原引发剂体系中的水溶性还原剂的实例包括例如 C,Brü FF6,Brü FF7或焦亚硫酸钠。
根据优选实施方案,使用水溶性氧化还原引发剂体系,例如叔丁基氢过氧化物,过氧化氢或过氧二硫酸的盐(例如过硫酸钠,过硫酸铵或过硫酸钾),与 C,Brü FF6,Brü FF7或焦亚硫酸钠结合。
根据本发明的优选实施方案,聚合反应由一种或多种水溶性自由基引发剂组成的引发剂体系引发。这意味着该引发剂体系排他地包含水溶性自由基引发剂,不包含任何在去离子水中在20℃和大气压下溶解度小于1wt%的引发剂。
专用水溶性引发剂是优选的,因为水不溶性引发剂通常在乳液聚合中并不十分有效,原因是其经由水相的迁移较差,以及在一些情况下还产生悬浮聚合,形成大颗粒,这对于某些应用而言是不希望有的。因此巨大的优点是本发明的方法允许显著更加经济地制备所需产物,而不损害其性能或特性,以及本发明的方法也可以为商业规模。
考虑到多相反应中水性聚合所常用的,可以适当选择本发明方法中使用的引发剂或引发剂体系的量。通常,基于待聚合的单体总量,使用的引发剂的量不超过5wt%。基于待聚合的单体总量,使用的引发剂的量优选为0.05至2.0wt%。
关于这一点,有可能在聚合开始时就将引发剂总量包括在反应器的初始投料中。另外,可以在初始投料中包括一部分引发剂,以及在已经引发聚合之后在一个或多个步骤中或连续地添加余量引发剂。添加可以单独进行,或与其它组分,例如乳化剂一起添加。根据优选实施方案,将引发剂缓慢加入到聚合反应过程中,例如速率为每分钟不超过待添加的引发剂总量的2%,或优选速率为每分钟不超过待添加的引发剂总量的1%。
由本发明方法获得的聚合物分散体的分子量可以通过添加少量的一种或多种转移剂,即分子量调节剂物质来加以调节。这些转移剂通常以基于待聚合的全部单体,至多2wt%的量使用。作为转移剂,可以使用本领域中已知的所有物质。优选例如有机硫代化合物,硅烷,烯丙醇,和醛类。
稳定体系
聚合和其后过程中,本发明方法获得的聚合物分散体可以以水性聚合物分散体或胶乳的形式稳定化。聚合物分散体因此优选在稳定体系存在下制备并且将包含稳定体系,该稳定体系通常包含乳化剂,特别是非离子型乳化剂和/或阴离子型乳化剂,和或保护胶体。也可以使用不同稳定剂的混合物。
使用的乳化剂的量优选为至少0.5wt%,基于聚合物分散体中的单体总量。通常,乳化剂可以以基于聚合物分散体中的单体总量至多约8wt%的量使用。非离子对阴离子型乳化剂的重量比可以在宽范围内变化,例如为1∶1至50∶1。优选的乳化剂包括具有环氧烷基团的非离子型乳化剂,和/或具有硫酸盐、磺酸盐、磷酸盐和/或膦酸盐基团的阴离子型乳化剂。如果需要,这种乳化剂可以与分子或分散水溶性聚合物,优选与聚乙烯醇一起使用。
合适的非离子型乳化剂的实例包括酰基,烷基,油烯基和烷基芳基乙氧基化物。这些产品是市售的,例如商品名Genapol,Lutensol或Emulan。它们包括例如乙氧基化单-、二-和三-烷基酚(EO度:3至80,烷基取代基:C4至C12)以及乙氧基化脂肪醇(EO度:3至80;烷基:C8至C36),特别是C10-C14脂肪醇(EO 3-80)乙氧基化物,C11-C15羰基法醇(EO 3-80)乙氧基化物,C16-C18脂肪醇(EO 3-80)乙氧基化物,C11羰基法醇(EO 3-80)乙氧基化物,C13羰基法醇(EO 3-80)乙氧基化物,具有20个环氧乙烷基团的聚氧化乙烯单油酸山梨醇酐酯,具有10wt%的最小环氧乙烷含量的环氧乙烷和环氧丙烷的共聚物,油醇的聚环氧乙烷(EO 3-80)醚,和壬基苯酚的聚环氧乙烷(EO 3-80)醚。特别合适的是脂肪醇,更具体地油醇,硬脂醇或C11烷基醇的聚环氧乙烷(EO 3-80)醚。
用于制备聚合物分散体的非离子型乳化剂的量通常为约1wt%至约8wt%,优选为约1wt%至约5wt%,更优选为约1wt%至约4wt%,基于单体总量。也可以使用非离子型乳化剂的混合物。
合适的阴离子型乳化剂的实例包括链长C12至C20的线性脂肪族羧酸的钠、钾和铵盐,羟基十八烷-磺酸钠,链长C12至C20的羟基脂肪酸的钠、钾和铵盐及其磺化和/或硫酸化和/或乙酰化产物,仲烷基磺酸盐,烷基硫酸盐,包括三乙醇胺盐形式的那些,烷基(C10-C20)磺酸盐,烷基(C10-C20)芳基磺酸盐,及其磺化产物,木质磺酸及其钙、镁、钠和铵盐,树脂酸,加氢和脱氢树脂酸,及其碱金属盐,十二烷基化二苯醚二磺酸钠,月桂基硫酸钠,EO度为1至10的硫酸盐化烷基或芳基乙氧基化物,例如乙氧基化月桂基醚硫酸钠(EO度3),或二酯,优选双-C4-C18烷基酯的盐,具有4至8个碳原子的磺化二羧酸的盐,或这些盐的混合物,优选琥珀酸酯的磺化盐,更优选磺化琥珀酸的双-C4-C18烷基酯的盐,例如碱金属盐,或聚乙氧基化烷醇或烷基酚的磷酸盐。
基于单体总量,使用的阴离子型乳化制的量优选为约0.1wt%至约3.0wt%,优选约0.1wt%至约2.0wt%,更优选约0.5wt%至约1.5wt%。也可以使用阴离子型乳化剂的混合物。
水性聚合物分散体可以进一步包含保护胶体作为稳定体系的一部分,该保护胶体优选为水溶性或胶状可分散聚合物。保护胶体可以基于纤维素醚,聚乙烯醇,聚乙烯基吡咯烷酮,聚氨酯基共聚物,聚丙烯酸,马来酸-苯乙烯共聚物或其它水溶性聚合物。由本发明方法获得的聚合物分散体中使用的适宜保护胶体包括基于纤维素醚的水溶性或水可分散聚合改性的天然物质。当以1wt%水溶液的形式在25℃测试时,这种纤维素醚具有约5至约5,000mPas,优选约10至约1,500mPas,更优选约10至约500mPas的粘度。纤维素醚的混合物可用来达到这些粘度值。合适的纤维素醚材料的实例包括甲基纤维素,羟乙基纤维素,羟丙基纤维素,羟丙基甲基纤维素,羟乙基纤维素乙基醚,甲基羟乙基纤维素及其组合。合适的纤维素醚材料可以商品名和获得。以商品名市售的羟乙基纤维素(HEC)是最优选的。
保护胶体,优选羟乙基纤维素和聚乙烯醇,可以例如由烷基,芳基,环烷基加以疏水改性,或例如由羧基,硫酸盐,磺酸基,羟基加以亲水改性。疏水改性的纤维素醚可以包含已经用长链烃基疏水改性来降低其水溶性的纤维素醚。此类疏水改性的纤维素醚为例如US 4,228,277,4,352,916和4,684,704中描述的那些。
保护胶体可以单独使用或组合使用。在组合的情况下,两种或多种胶体各自可以分子量不同,或它们可以分子量以及化学组成不同,例如在聚乙烯醇的情况下,水解度不同。
优选的保护胶体为羟乙基纤维素和聚乙烯醇。合适的聚乙烯醇具有60至100mol%的水解度,和在20℃下4%水溶液具有2至70mPas,特别是4至40mPas的粘度。
当使用保护胶体时,基于使用的单体总量,其量通常为0.1至10wt%,优选0.2至8wt%,甚至更优选0.4至6wt%。
在优选实施方案中,本分散体完全不含保护胶体,或基于使用的单体总量,保护胶体的量小于1wt%,更优选小于0.7wt%。
除共聚物乳液聚合期间使用的乳化剂和保护胶体之外,在聚合之后也可添加其它乳化剂,保护胶体和/或其它稳定剂作为后添加剂。
(共)聚合物分散体制备
可以使用乳液聚合步骤制备包含本发明方法获得的水性(共)聚合物的(共)聚合物分散体,所述乳液聚合步骤导致制备水性胶乳形式的聚合物分散体。此类水性聚合物分散体的制备是公知的,已经在许多场合描述,因此是本领域已知的。这些步骤例如在US 5,849,389,以及在Encyclopedia of Polymer Science and Engineering,8卷,659页(1987)中描述。
聚合可以以任何已知的方式,用不同的单体组合以一步、两步或多步进行,产生具有均相或非均相,例如核壳或半球形态的颗粒的聚合物分散体。但是为了产生非均相颗粒形态,也可以使用其它单体情况,并且这是本领域技术人员已知的。在一个优选实施方案中,通过聚合第一单体或第一单体混合物形成第一种聚合物,随后在该第一种聚合物上聚合第二单体或第二单体混合物,产生具有非均相形态的共聚物。
可以使用任何合适的反应器系统。聚合可以例如由间歇或半间歇进行,即其中所有单体提前添加,或其中在聚合反应过程中缓慢添加部分单体。
可以将单体连续地,逐渐地或作为使用的单体总量的单一进料添加量的形式加入到聚合容器中。单体可以以纯净单体的形式使用,或者可以以预混合乳液的形式使用。单体可以一起计量,或者以单独的进料流计量。乙烯作为共聚单体,可以通过将其泵送(或添加/进料)进入聚合容器并使其保持在适当压力下而在任何步骤添加。为了控制乙烯添加量,可以使用恒定压力或恒定进料速率添加乙烯。也可将两种方法组合,例如开始使用恒定压力添加,然后在一定聚合时间之后,应用恒定进料速率,或反之亦然。
根据本发明的优选实施方案,最初进料基于待共聚合的主要单体总量至多100%的主要单体,余量在聚合反应阶段过程中连续添加。这包含单体的完整预先进料以及仅部分待(共)聚合单体的初始进料。根据本发明的更优选实施方案,基于全部单体,使用的一部分单体,优选至多约50wt%,更优选约1wt%至约50wt%,甚至更优选约5wt%至约30wt%被以初始进料形式引入,以引发聚合,并且余量单体在聚合反应阶段期间计量加入反应混合物中。
此外,在某些实施方案中有利的是设定聚合开始时的特定粒度和粒度分布,以进行种子聚合或在初始进料中包括单独制备的种子。当本发明的方法中使用种子胶乳时,其在乳液聚合开始之前使用,例如为分散体的约0.5至约15wt%,优选分散体的约1至约10wt%。
混合若干聚合成分,即乳化剂,单体,引发剂,保护胶体等的方法可以广泛变化。通常可以在聚合容器中最初形成含有至少一些乳化剂的水性介质,随后向容器中添加各种其它聚合成分。当使用时,用于稳定和/或保护胶体的乳化剂可以在聚合开始时完全引入初始进料中,或者可以在初始进料中包括部分乳化剂以及部分计量加入,或者在聚合进行期间将全部量计量加入。优选的是在初始进料中包括全部乳化剂或大部分乳化剂。
本发明的乳液聚合优选至少部分在压力下进行。聚合通常在适当压力,优选2至150bar,更优选5至100bar,甚至更优选10至85bar下进行。
当在本发明的方法中执行压力步骤时,可以仅在反应混合物中10wt%至50wt%的液态主要单体已经转化之后开始计量气态单体,由此形成非均相颗粒形态。当本发明的方法使用压力步骤操作时,另外也可以在乳液聚合开始时开始计量气态单体,使得与液态单体一起发生转化,形成均相颗粒形态。
在涉及例如水性共聚物分散体的典型聚合方法中,单体,例如乙酸乙烯酯,乙烯和其它单体可以在一种或多种引发剂,至少一种乳化剂和保护胶体组分存在下,在至多120bar的压力下在水性介质中聚合。聚合容器中的水性反应混合物可以借助于合适的缓冲剂保持在约2至约7的pH。
根据本发明的优选实施方案,使50至99%的乙酸乙烯酯,1至40%的乙烯,和任选至多30%的一种或多种其它主要单体和/或至多10%的一种或多种辅助单体共聚。在这种情况下,优选的是在聚合反应阶段开始时,将乙烯压力升高到总计为待共聚的乙烯总量的至少30%,优选至少60%的值,然后聚合反应阶段持续时间的至多约50%至60%,将乙烯压力保特在总计为待共聚的乙烯总量的至少50%的值,以及随后不进一步添加,使得反应容器中的残留乙烯聚合导致乙烯压力连续降低。在这种情况下,优选通过施加预定乙烯压力而不是通过控制进料速率来控制乙烯的添加。
后聚合和进一步处理
在要求保护的乳液聚合方法结束之后可以去除残余单体。这可以使用已知的方法完成。如以上已经解释的,基于全部单体,残余单体通常不超过约2wt%,优选不超过约1wt%,特别优选不超过约0.5wt%。因此,后聚合反应可以任选接着要求保护的乳液聚合方法之后进行,其中在基于全部单体,多至98wt%,优选多至99wt%,特别优选多至99.5wt%的单体已经聚合之后,后聚合反应开始。
后聚合可以在与结束温度相同的温度或者更低的温度下进行。后聚合阶段之后通常为冷却阶段。后聚合可以包括优选在不同反应器中进行的最终氧化还原处理。这种最终氧化还原处理并非上述聚合反应的一部分。
因此,本发明还涉及一种用于使自由基聚合性烯属不饱和主要单体和任选可与之共聚的其它辅助单体乳液聚合的方法,其制备具有低残留单体含量的聚合物,该方法包括上述乳液聚合的方法,和随后的后聚合反应。
也可以通过蒸馏,优选在减压下蒸馏,和适当时,用穿过产物或在产物之上通过的惰性夹带气体,例如空气,氮气或蒸汽来去除挥发性残余单体。去除残余单体的方法例如由WO-A-04/22609已知。
聚合或后聚合之后,可以通过添加水或通过蒸馏去除水,将所得水性聚合物分散体的固含量调节至所需水平。通常,聚合之后所需的聚合物固含量水平为约20wt%至约70wt%,优选约30wt%至约65wt%,更优选约40wt%至约60wt%,基于聚合物分散体总重量。
本发明制备的聚合物分散体的pH通常为2至8,优选为4至7。
平均粒度通常为小于1.2μm,优选小于1,000nm,和更优选小于400nm(如下所述通过激光衍射或激光气溶胶光谱测定,取决于颗粒的尺寸)。
根据本发明制备的聚合物分散体的粘度通常为10mPas至50,000mPas,优选为100mPas至20,000mPas,和更优选为100至15,000mPas(通过布氏粘度计,在23℃,20rpm和正确测量范围的对应转轴下测量)。
聚合物分散体的用途
本发明也涉及一种水性(共)聚合物分散体,其包含至少一种由本发明方法形成的(共)聚合物。
本发明的聚合物分散体可以用于通常的应用领域。
本发明的聚合物分散体可以用作任何所需基材的粘结剂,例如用作基材涂料(特别是油漆)的含颜料水性制备品,地毯背涂层,纸张(特别是纸饱和和纸张涂层),粘合剂,或织物(特别是印染和织物饰面)和无纺布(工程织物)的粘结剂。另外,本发明的水性共聚物分散体可以例如用作木纤维板或人造革的粘结剂,由纸张纤维或聚合物纤维制造的绝缘材料的粘结剂,以及用作优选用于多孔组分的乳液修饰剂和釉料,密封化合物和密封组合物的粘结剂。
根据本发明的特别优选实施方案,本发明的分散体用作油漆的粘结剂,所述油漆例如内用和/或外用乳胶漆,特别是低挥发内用漆,低挥发外用漆,或超过临界颜料体积浓度(PVC)的油漆配制料。“低挥发”油漆具有与总挥发性有机化合物(TVOC)有关的低挥发性(根据用于聚合物分散体TVOC含量测量的ISO11890-2法;见下文)。用本文所述水性共聚物分散体制备的涂料通常将含有基于涂料总重量,按重量计低于2,000ppm,优选低于1,000ppm,更优选低于500ppm,和甚至更优选低于100ppm的TVOC,根据ISO 11890-2。
根据本发明的水性共聚物分散体优选具有低于100ppm,更优选低于30ppm,和甚至更优选低于10ppm的甲醛含量,如根据用于涂料组合物的VdL-RL03测试法(VdL Guideline 03),由乙酰基-丙酮法测定。
上述共聚物分散体可以与填料材料,外加水和/或任何任选的其它成分,例如一种或多种助剂结合,形成本文的水性涂料组合物。如此形成的水性组合物的固含量通常将为全部组合物的约30wt%至约75wt%。更优选,本文水性涂料组合物的固含量将为全部组合物的约40wt%至约65wt%。这些应理解为表示除了水,但至少有固体粘结剂,填料,颜料,增塑剂和聚合物助剂总量的制备品的所有成分。
可以使用的颜料为用于所述预定用途的本领域技术人员已知的所有颜料。用于本发明水性制备品,优选用于乳胶漆的优选颜料例如为二氧化钛,优选金红石形式的二氧化钛,硫酸钡,氧化锌,硫化锌,碱式碳酸铅,三氧化锑和锌钡白(硫化锌和硫酸钡)。水性制备品也可以含有彩色颜料,例如氧化铁,炭黑,石墨,发光颜料,锌黄,锌绿,群青,锰黑,硫化锑,锰紫,巴黎蓝或巴黎绿。除无机颜料之外,根据本发明的制备品也可以含有有机彩色颜料,例如墨鱼墨,藤黄,卡塞尔褐土,甲苯胺红,对位红,汉萨黄,靛青,偶氮染料,蒽醌和靛类染料以及二嗪,和喹吖啶酮,酞菁,异吲哚啉酮和金属络合物颜料。
可以使用的填料为用于所述预定用途的本领域技术人员已知的所有填料。优选的填料为铝硅酸盐,例如长石,硅酸盐,例如高岭土,滑石,云母,菱镁矿,碱土金属碳酸盐,例如碳酸钙,例如方解石或白垩形式,碳酸镁,白云石,碱土金属硫酸盐,例如硫酸钙,以及二氧化硅。填料可以单一组分或填料混合物的形式使用。填料混合物,例如碳酸钙/高岭土以及碳酸钙/滑石,是实践中优选的。合成树脂粘结底灰也可以含有较粗聚集体,例如砂子或砂岩颗粒。通常,细碎填料是乳胶漆中优选的。
为了提高遮盖力和节约白色颜料,乳胶漆中优选经常使用细碎填料,例如沉淀碳酸钙或不同粒度的不同碳酸钙的混合物。彩色颜料和填料的混合物优选用于调节色调的遮盖力以及色浓度。
在一个实施方案中,涂料组合物可以包含30至90%的至少一种填料,0.1至25%的至少一种颜料,和5至60%,优选5至20%的本发明的水性(共)聚合物分散体。
常用的助剂包括润湿剂或分散剂,例如多磷酸钠、钾或铵,聚丙烯酸和聚马来酸的碱金属和铵盐,聚膦酸盐,例如1-羟基乙烷-1,1-二膦酸钠,和萘磺酸盐,特别是其钠盐。另外,合适的氨基醇,例如2-氨基-2-甲基丙醇可以用作分散剂。分散剂或润湿剂优选以基于乳胶漆总重量,0.1至2wt%的量使用。
此外,助剂也可以包含增稠剂,例如纤维素衍生物,例如甲基纤维素,羟乙基纤维素和羧甲基纤维素,和酪蛋白,阿拉伯树胶,黄蓍胶,淀粉,海藻酸钠,聚乙烯醇,聚乙烯吡咯烷酮,聚丙烯酸钠,基于丙烯酸和(甲基)丙烯酸的水溶性共聚物,例如丙烯酸/丙烯酰胺和(甲基)丙烯酸/丙烯酸酯橡胶,以及所谓的缔合增稠剂,例如苯乙烯/马来酸酐聚合物或优选本领域技术人员已知的疏水改性聚醚氨酯(HEUR),疏水改性丙烯酸共聚物(HASE)聚醚多元醇。也可以使用无机增稠剂,例如膨润土或锂皂石。增稠剂优选以基于水性制备品总重量的0.1至3wt%,特别优选0.1至1wt%的量使用。
另外,基于链烷烃和聚乙烯的蜡,以及消光剂,防沫剂,防腐剂和拒水剂,杀菌剂,纤维和本领域技术人员已知的其它添加剂也可以用作本发明的水性制备品中的助剂。
本发明的分散体不仅可用于制造无溶剂和增塑剂制备品,而且可用于制造含有溶剂和/或增塑剂作为成膜助剂的涂料体系。成膜助剂通常是本领域技术人员已知的,通常可以以基于制备品中存在的(共)聚合物,0.1至20wt%的量使用,使得水性制备品具有低于15℃,优选为0℃至10℃的最小成膜温度。考虑到本发明的(共)聚合物分散体的有益性能,通常并非必须使用这些成膜助剂。在优选的实施方案中,本发明的水性制备品因此不含有成膜助剂。在不添加成膜剂的基础上,涂料组合物可以具有小于或等于5℃的最小成膜温度。
本发明的水性制备品是稳定的流体体系,可以用于涂布许多基材。因此,本发明也涉及涂布基材的办法和涂料材料本身。合适的基材为例如木材,混凝土,矿物基材,金属,玻璃,陶瓷,塑料,底灰,墙纸,纸张,以及涂布、涂底漆或风化的基材。对待涂布的基材应用该制备品以依赖于制备品形成的方式起作用。根据制备品的粘度和颜料成分以及根据基材,应用可以通过辊涂,刷涂,刮涂或喷涂完成。
本发明的含颜料水性制备品的颜料体积浓度(PVC)通常为高于5%,优选为10至90%。在涂料组合物中使用水性共聚物分散体的情况下,水性共聚物分散体的特殊特征为即使在高颜料体积浓度(PVC)下,即在超过临界PVC的高度填充配制的组合物中,也能够具有很好的抗湿擦性。因此,在特别优选实施方案中,PVC为60至90%,特别是70至90%。发明的乳液同样适用于低PVC油漆,例如缎光漆和半光漆的介质。在这种情况下,PVC为10至45%。
此外,本发明的聚合物分散体可以用作任何所需基材的粘合剂,优选用于粘合多孔和半多孔基材。这些应用包括例如粘结纸张,卡片,瓦楞纸板,泡沫材料,水泥,皮革,织物或挤压层压材料,适当时与无孔基材,聚合物薄膜结合。优选的粘合剂粘结的实例为纸张/纸张或纸张/聚合物薄膜组合。其它优选的粘合剂粘结为聚合物薄膜/聚合物薄膜组合。
分散体可以以其原本形式使用,或者以可水再分散性分散体粉末的形式使用。对于改善的再分散性,优选添加保护胶体,例如聚乙烯醇和/或纤维素醚,和/或合适的防结块剂。这些粉末可以通过分散体的喷雾干燥,以常规方式获得。
本发明的分散体或分散体粉末可以例如用于建筑物产品,与水硬性粘结剂、水泥结合,实例为波特兰水泥,铝酸盐,熔渣,氧化镁,和/或磷酸盐水泥,石膏和水玻璃,用于制造建筑物粘合剂,特别是瓷砖粘合剂和用于集成的绝热系统、壁或天花板的粘合剂,底灰,抹平胶料,地板胶料,家具箔片,找平胶料,灌浆,接缝灰浆,和石膏。
特别优选使用本发明的水性(共)聚合物分散体作为油漆、纸张饱和和纸张涂料、粘合剂、无纺布、织物、地毯背胶、建筑物或粉末中的粘结剂。
以下实施例用来说明本发明,但并不限制的本发明范围。
实施例
共聚物分散体粒度测量
本文使用的共聚物分散体内的固体颗粒尺寸由激光气溶胶光谱(LAS)测定。该LAS法在出版物Kunstharz Nachrichten 28;“Characterization and Quality Assurance of PolymerDispersions”;Oktober 1992,Dr.J.Paul Fischer中描述。该办法使用由Spectra Physics提供的Nd:YV04激光器(Millenia II),激光功率为2W,波长为532nm。测试仪为由Burle(前身RCA)提供的Bialkali Photocathode Typ 4517。在40°检测喷雾干燥的单个颗粒的散射光。用TMCA的1024通道的多通道分析仪评价数据。,在100ml去离子和过滤水(导电性为18.2μS/m)中稀释0.2ml的分散体试样,用于粒度测量。将试样在Beckmann喷口之上喷雾干燥,以及用氮气干燥。用β射线(Kr-85)中和单个颗粒,然后由单个颗粒激光器散射进行研究。通过应用该办法,获得80nm至550nm范围内的数目和质量平均值,以及平均粒度值dn、dw、dz和dw/dn。
实施例8至15中使用的共聚物分散体中的固体颗粒尺寸通过激光器衍射,使用市售Beckmann Coulter LS 13320设备测定,和该方法和设备的详细说明可获自Beckman Coulter。对于实际测量,在5ml水中稀释大约5滴每个试样(取决于实际粒度)。充分混合之后,将稀释液转移进测量容器中。由该设备自动进一步稀释试样,以产生对于方法和设备最佳的衍射强度。以20kHz,70W使用1min超声波处理。为测量小颗粒,使用PIDS(极化强度差示散射)。通过应用该办法,获得0.017-2,000μm范围内的数目和质量平均值,以及平均粒度值dn、dw、dz和dw/dn。
共聚物玻璃化转变温度(Tg)测量
使用商业差示扫描量热计Mettler DSC 820,以10K/min测定玻璃化转变温度Tg。为了评价,使用第二加热曲线和计算DIN中点。
共聚物分散体挥发性有机化合物/乙酸乙烯酯单体含量(DIN ISO 11890-2)
使用ISO 11890-2测试法测定共聚物分散体的总挥发性有机化合物含量。该办法通过直接注入毛细管气相色谱柱,测定挥发性有机组分(VOC)或残余单体,例如乙酸乙烯酯的残留水平。使用的办法按照DIN ISO 11890-2指示,其中总挥发性有机组分(TVOC)定义为沸点低于十四烷的所有挥发性有机组分的总和。该组分具有253℃的沸点。使用装有PPC(气动压力控制)的Perkin Elmer气相色谱仪(AutoSystem XL),其具有Varian柱V624,60米,320μm内径和1.8μm薄膜厚度。载体气体为H2。测试仪为FID。对于试样制备,使用Gilson Micromann 250正排量滴管,将大致150μl试样放入已称皮重的管瓶。称量自动采样器管瓶(g),结果记录为因子值(divisor value)。将大致1.5ml稀释剂溶液(去离子水中含有100ppm的甲基异丁酮(MIBK),作为内标物)加入到自动采样器管瓶中。称量自动采样器管瓶(g),结果记录为乘子(multiplier)。使用涡流混合器将自动采样器管瓶充分混合,直到管瓶中的溶液完全均相。然后将试样管瓶放置在气相色谱仪的试样转盘上,按照ISO 11890-2测量。最初校准各个单一VOC/残余单体值。结果为所有单一VOC值的总和,单一VOC值为TVOC参数或仅为以ppm计的单个残余单体。
Fikentscher K值
Fikentscher K值范围(H.Fikentscher,Cellulosechemie 13(1932),58-64和71-74)为特性粘度的量度,类似于DIN 53726,以及为聚合物分子量的指示。通常K值升高与较高强度和刷洗性提高有关。为测定K值,将1g当量的干燥聚合物(=2g的50%含固体分散体)在室温下溶于100ml二甲基甲酰胺(DMF),同时搅拌(直到完全溶解,至少1h),使用Mikro-Ostwald-Viscometer(毛细管类型518 13/Ic[2ml];Schott AVS 400+CT1150)测定23℃下的溶液粘度。然后,在相同的粘度计中测定23℃下100ml DMF中1g水的粘度。将粘度读数和聚合物浓度代入以下公式(A),计算k值。将k值乘以1000得到K值。
公式(A):logηc/ηo=[(75*k2)/(1+1.5k*c)+k]*c
以下记述四组彼此独立的实施例A至D。每组实施例A至D含有至少一个根据本发明的实施例和至少一个对比例。由聚合反应过程中添加的单体量,在以下实施例中计算起始温度、结束温度和聚合温度升高期间的总转化率。
实施例A
实施例A-1-VAE-基共聚物分散体制备
在装有锚式搅拌器(以150rpm运转),加热夹套,计量泵并具有30升容积的压力反应器中添加以下组分的水基溶液:
10245g水(去离子)
164g去离子水中聚乙烯醇溶液(29%),即部分水解[88水解度(mol%)],在20℃形成4.50cP±0.50的4%溶液粘度。
563g的C11烷基聚乙二醇醚(28mol环氧乙烷)-非离子型乳化剂(去离子水中70%)。
234g的C11烷基聚乙二醇醚(7mol环氧乙烷)硫酸钠-阴离子型乳化剂(去离子水中30%)。
222g乙烯基磺酸钠(去离子水中30%)。
33.3g乙酸钠(无水)
5.25g偏亚硫酸氢钠
0.0302g的Mohr氏盐(Fe2+盐)
1.59g消泡剂Agitan 282
将聚乙烯醇(29%)在90℃溶于去离子水2小时。用氮气吹扫反应器去除氧气。从总量11551g乙酸乙烯酯中将10%的乙酸乙烯酯加入到反应器中的水相。打开乙烯阀,在环境温度下将反应器增压至30bar(25℃下大约56%的乙烯阶段1),然后再次关闭(乙烯总量:1575g)。
反应器温度上升至75℃。60℃(聚合阶段起始温度)下,将引发剂进料,过二硫酸钠(813g去离子水中19.3g)(以大约380g/h速率经60min)加入反应器。65℃下,经60min将其余乙酸乙烯酯(90%)与65.6g甲基丙烯酸缩水甘油酯一起加入反应器。在相同温度(65℃)下,再次打开乙烯阀,在75℃、56bar的最大压力下经约10min将剩余乙烯(44%)加入反应器。当添加完乙烯(75℃)并且反应显示放热行为时,通过升高夹套温度和/或使用聚合热使反应器温度上升至115℃。当单体进料(VAM)完成时,将引发剂速率升高至1200g/h的最大速率。引发剂进料完成(聚合阶段终点)之后,反应温度在85℃保持30min。然后将反应器冷却至大致40℃,并排出批料。此时通过引入Brüggolit FF 6(亚磺酸衍生物的钠盐,得自L.Brueggemann KG)(126g去离子水中13g)和随后引入Trigonox AW 70(28g),进行最终的氧化还原处理。排料之前将产物搅拌30min。
实施例A-2-VAE-基共聚物分散体制备
配方和步骤与实施例1相同,但是温度升高到120℃(结束温度)。
实施例A-3(对比)-VAE-基共聚物分散体制备
在装有锚式搅拌器(以150rpm运转),加热夹套,计量泵并具有30升容积的压力反应器中添加以下组分的水基溶液:
10295g水(去离子)
454g去离子水中聚乙烯醇溶液(29%),即部分水解[88水解度(mol%)],在20℃形成4.50cP±0.50的4%溶液粘度。
564g的C11烷基聚乙二醇醚(28mol环氧乙烷)-非离子型乳化剂(去离子水中70%)。
223g乙烯基磺酸钠(去离子水中30%)。
33.5g乙酸钠(无水)
5.27g偏亚硫酸氢钠
0.0303g的Mohr氏盐(Fe2+盐)
1.59g消泡剂Agitan 282
将聚乙烯醇(29%)在90℃溶于去离子水2小时。用氮气吹扫反应器去除氧气。从总量11590g乙酸乙烯酯中将25%的乙酸乙烯酯加入到反应器中的水相。打开乙烯阀,在环境温度下将反应器增压至30bar(25℃下大约59%的乙烯阶段1),然后再次关闭(乙烯总量:1580g)。反应器温度上升至80℃。80℃(聚合阶段起始温度)下,将引发剂进料,过二硫酸钠(817g去离子水中19.4g)(以大约970g/h速率经3-4min)加入反应器,直到反应开始。85℃下,经50min将其余乙酸乙烯酯(75%)与65.9g甲基丙烯酸缩水甘油酯一起加入反应器。在相同温度(85℃)下,再次打开乙烯阀,在105℃、67bar的最大压力下经约10min将剩余乙烯(41%)加入反应器。通过控制引发剂速率,将聚合温度保持在大约110℃。当单体进料(VAM)完成时,将引发剂速率升高到1200g/h的最大速率,将夹套温度保持在95℃,直到通过减慢反应降低反应器温度。引发剂进料完成(聚合阶段终点,100℃)之后,反应温度在85℃保持30min。然后将反应器冷却至大致40℃,并排出批料。此时通过引入Brüggolit FF 6(亚磺酸衍生物的钠盐,得自L.Brueggemann KG)(126g去离子水中13g)和随后引入Trigonox AW 70(28g),进行最终的氧化还原处理。排料之前将产物搅拌30min。
表1:实施例A-1至A-3的工艺参数
表2:实施例A-1至A-3中获得的VAE共聚物分散体的性能
根据表2,K值为聚合物分子量的量度,显示与恒定高温下相同单体缓慢添加时间下的聚合方法相比,本发明方法的值高。较高的K值导致抗湿擦性更好,内聚力和耐热性更好。
实施例B
实施例B-1(对比)-VAE-基共聚物分散体制备
在装有锚式搅拌器(以150rpm运转),加热夹套,计量泵并具有70升容积的压力反应器中添加以下组分的水基溶液:
25934g水(去离子)
1478g的C11烷基聚乙二醇醚(28mol环氧乙烷)-非离子型乳化剂(去离子水中70%)。
1154g十二烷基硫酸钠(去离子水中15%)
591g乙烯基磺酸钠(去离子水中30%)
88.6g乙酸钠(无水)
14.1g偏亚硫酸氢钠
0.081g的Mohr氏盐(Fe2+盐)
4.24g消泡剂Agitan 282
用氮气吹扫反应器去除氧气。从总量31014g乙酸乙烯酯中将5%的乙酸乙烯酯加入到反应器中的水相。打开乙烯阀,在环境温度下将反应器增压至36bar(25℃下大约60%的乙烯阶段1),然后再次关闭(乙烯总量:4299g)。反应器温度上升至65℃。60℃(聚合反应阶段起始温度)下,将引发剂进料,过二硫酸钠(1236g去离子水中115g)加入(引发剂1:33.3%,经135min)反应器。65℃下,经242min将其余乙酸乙烯酯(95%)加入反应器。在相同温度(65℃)下,再次打开乙烯阀,在65℃、36bar的最大压力下经约150min将剩余乙烯(40%)加入反应器。当添加完引发剂1时,立即继续引发剂2(33.3%,经90min)。添加完引发剂2之后,继续引发剂3(33.4%,经60min)。29min之前停止乙酸乙烯酯进料,经29min使反应器温度升高至85℃,并保持60min(该温度下28min之后,引发剂进料完成(聚合反应阶段终点))。然后将反应器冷却至大致40℃,并排出批料。此时通过引入Br ü ggolit FF 6(亚磺酸衍生物的钠盐,得自L.Brueggemann KG)(250g去离子水中26g)和随后引入Trigonox AW 70(60g),进行最终的氧化还原处理。排料之前将产物搅拌30min。
实施例B-2-VAE-基共聚物分散体制备
在装有锚式搅拌器(以150rpm运转),加热夹套,计量泵并具有70升容积的压力反应器中添加以下组分的水基溶液:
25934g水(去离子)
1478g的C11烷基聚乙二醇醚(28mol环氧乙烷)-非离子型乳化剂(去离子水中70%)。
1154g十二烷基硫酸钠(去离子水中15%)
591g乙烯基磺酸钠(去离子水中30%)
88.6g乙酸钠(无水)
14.1g偏亚硫酸氢钠
0.081g的Mohr氏盐(Fe2+盐)
4.24g消泡剂Agitan 282
用氮气吹扫反应器去除氧气。从总量31014g乙酸乙烯酯中将40%的乙酸乙烯酯加入到反应器中的水相。反应器温度升高至50℃。打开乙烯阀,在50℃下将反应器增压至45bar(100%乙烯),然后再次关闭(乙烯总量:4299g)。在30℃(聚合反应阶段开始),在10min内将引发剂I,过二硫酸钠(618g去离子水中78g)加入反应器。在50℃和放热行为下,将单体进料(60%乙酸乙烯酯)经118min加入反应器。相同时间(50℃反应器温度)下在62min内从50开始升温至85℃。然后将反应器温度保持在85℃另外56min。90min之后(乙酸乙烯酯进料开始之后),在28min内添加引发剂II溶液,过二硫酸钠(618g去离子水中37g)。所有进料(乙酸乙烯酯和引发剂II溶液)完成(聚合反应阶段终点)之后,将反应器温度在85℃保持另外60min。然后将反应器冷却至大致40℃,并排出批料。此时通过引入Br ü ggolit FF 6(亚磺酸衍生物的钠盐,得自L.Brueggemann KG)(250g去离子水中26g)和随后引入Trigonox AW 70(60g),进行最终的氧化还原处理。排料之前将产物搅拌30min。
表3:实施例B-1和B-2的工艺参数
FLAT漆(51PVC)的制备(参见US 2010/0056696的表3)。
粘结剂含量(分散体):29%
表4:
FLAT漆(51PVC)的抗擦性测试ASTM D2486,根据US2010/0056696,[0158]段。
表5:对比和本发明VAE共聚物分散体的结果
在甚至显著更短的聚合时间下,与对比例B-1相比,根据本发明的实施例B-2显示,无光漆中的K值更高和抗湿擦性更好。
实施例C
实施例C-1至C-5(对比)-恒定聚合温度下VAE基共聚物分散体的制备:
在装有锚式搅拌器(以220rpm运转),加热夹套,计量泵,用于控制添加量和反应器温度的DCS(分布控制系统)并具有29.2升容积的压力反应器中添加以下组分:
10168g水(去离子)
672g烷基聚乙二醇醚(C11烷基-30mol的环氧乙烷,浓度65%)
376g烷基苯磺酸盐(浓度20%)
251g乙烯基磺酸钠(30%)
37.6g乙酸钠(无水)
3.01g的Brueggolite FF6
0.62g的Mohr氏盐
1.51g的EDTA四钠盐
6.17g的Agitan 282
用氮气吹扫反应器去除氧气。从总量13185g乙酸乙烯酯中将初始量的乙酸乙烯酯加入到反应器中的水相。打开乙烯阀,用840g乙烯将反应器增压。将557g去离子水中12.0g的Brueggolite FF6的10%溶液(还原剂溶液)并联加入反应器,使反应器温度上升至45℃并使系统平衡。为引发和保持反应,将71%的上述Brueggolite FF6水溶液和557g去离子水中30.1g过硫酸钠的77%溶液(氧化剂溶液)并联和经210min(聚合反应阶段开始)加入反应器。一旦反应器温度已经分别达到50℃(实施例C-1和C-4),85℃(实施例C-2和C-5)和67.5℃(实施例C-3),开始添加单体。为此,将剩余乙酸乙烯酯与75.26g乙烯基三甲氧基硅烷混合并在200min内加入到反应器中。以相同时间将1026g乙烯加入反应器。为了添加乙烯,借助于自动控制阀将反应器压力固定在30bar。一旦加入全部乙烯量,则关闭阀并使反应器压力降低,直到反应完成。
一旦已经将上述量的单体,乙烯,氧化剂和还原剂加入反应器中,聚合时期开始,其中在40min内添加剩余量的氧化剂和还原剂溶液。引发剂进料完成之后,聚合反应阶段到达终点。在此期间,通过反应器夹套使反应器温度升高或保持在85℃(结束温度)。实施例C-1至C-5的工艺参数在表6中总结。表6和以上说明中举出的聚合阶段持续时间的偏差针对C-1-C-5,原因是实际泵速与DCS中的给定值之间的偏差。对于所有实施例,上述210min和40min用作引发剂进料持续时间的DCS给定值。乳液的特性在表7中总结。
实施例C-6至C-8
使用温度陡升制备VAE基共聚物分散体:在装有锚式搅拌器(以220rpm运转),加热夹套,计量泵,用于控制添加量和反应器温度的DCS并具有29.2升容积的压力反应器中添加以下组分:
10168g水(去离子)
672g烷基聚乙二醇醚(C11烷基-30mol的环氧乙烷,浓度65%)
376g烷基苯磺酸盐(浓度20%)
251g乙烯基磺酸钠(30%)
37.6g乙酸钠(无水)
3.01g的Brueggolite FF6
0.62g的Mohr氏盐
1.51g的EDTA四钠盐
6.17g的Agitan 282
用氮气吹扫反应器去除氧气。从总量13185g乙酸乙烯酯中将初始量的乙酸乙烯酯加入到反应器中的水相。打开乙烯阀,用840g乙烯将反应器增压。将557g去离子水中12.0g的Brueggolite FF6的10%溶液(还原剂溶液)并联加入反应器,使反应器温度上升至45℃并使系统平衡。为引发和保持反应,将71%的上述Brueggolite FF6水溶液和557g去离子水中30.1g过硫酸钠的77%溶液(氧化剂溶液)并联和经210min(聚合反应阶段开始)加入反应器。一旦反应器温度已经达到升温的起始温度(实施例C-6:50℃,实施例C-7:67.5℃,实施例C-8:50℃),开始添加单体。为此,将剩余乙酸乙烯酯与75.26g乙烯基三甲氧基硅烷混合并在200min内加入到反应器中。以相同时间将1026g乙烯加入反应器。为了添加乙烯,借助于自动控制阀将反应器压力固定在30bar。一旦已经加入全部乙烯量,则关闭阀并使反应器压力降低,直到反应完成。在将乙酸乙烯酯/乙烯基三甲氧基硅烷混合物加入反应器的同时,使反应温度从升温的起始温度升高至85℃。一旦已经将上述量的单体,乙烯,氧化剂和还原剂加入反应器中,聚合时期开始,其中在40min内添加剩余量的氧化剂和还原剂溶液。引发剂进料完成之后,聚合反应阶段到达终点。在此期间,使反应器温度升高或保持在85℃(结束温度)。实施例C-6至C-8的工艺参数在表6中总结。C-8在初始210min添加时期及其后40min时期之间的引发剂进料中具有15min停止。对于所有实施例,上述210min和40min用作引发剂进料持续时间的DCS给定值。乳液的特性在表7中总结。
表6:实施例C-1至C-8的工艺参数
表7:实施例C-1至C-8的乳液的特性
与根据实施例C-1至C-8,由在高温下进行的聚合获得的产物相比,根据以上实施例C-6至C-8,由使用升温的方法的聚合反应获得的聚合物显示显著更高的K值(50&67.5℃相比于85℃;50℃相比于67.5℃和67.5℃相比于85℃)。
实施例D
实施例D-1(对比)-VAE-基共聚物分散体制备
在装有锚式搅拌器(以150rpm运转),加热夹套,计量泵并具有30升容积的压力反应器中添加以下组分的水基溶液:
10322g水(去离子)
173g,去离子水中的聚乙烯醇溶液(15%),即部分水解[88水解度(mol%)],在20℃形成4.50cP±0.50的4%溶液粘度。
557g的C11烷基聚乙二醇醚(28mol环氧乙烷)-非离子型乳化剂(去离子水中70%)
234g十二烷基硫酸钠(15%)
223g乙烯基磺酸钠(去离子水中30%)
32g乙酸钠(无水)
5.1g偏亚硫酸氢钠
0.03g的Mohr氏盐(Fe2+盐)
用氮气吹扫反应器去除氧气。从11856g乙酸乙烯酯和52g乙烯基三甲氧基硅烷组成的总量11908g单体混合物中将29%的混合物加入到反应器中的水相。打开乙烯阀,用149g乙烯(全部乙烯的13%)将反应器增压,然后再次关闭(乙烯总量:1144g)。
反应器温度上升至40℃。在该温度下,将引发剂进料,过二硫酸钠(715g去离子水中40g)以大约190g/h的速率经245min加入(聚合反应阶段开始)。根据升温可以清楚地看到聚合开始,该升温在25min内持续至65℃。达到65℃,在2个阶段中开始添加剩余的单体混合物:1.在20min内添加3.5%的单体混合物,2.在另外200min内添加剩余的67.5%。随着第二阶段开始,经60min添加剩余乙烯(995g,87%)(达到大约48bar的最大压力)。添加完成液态单体(添加的全部液态单体的80%)之前30min,在30min内将温度从65℃升高至85℃(结束温度),因此伴随添加结束(单体混合物和引发剂);即在聚合反应阶段的结尾,达到最终温度。将反应器在85℃保持另外60min,然后冷却至大致40℃,排出批料。
实施例D-2-VAE-基共聚物分散体制备
在装有锚式搅拌器(以150rpm运转),加热夹套,计量泵并具有30升容积的压力反应器中添加以下组分的水基溶液:
10322g水(去离子)
173g,去离子水中的聚乙烯醇溶液(15%),即部分水解[88水解度(mol%)],在20℃形成4.50cP±0.50的4%溶液粘度。
557g的C11烷基聚乙二醇醚(28mol环氧乙烷)-非离子型乳化剂(去离子水中70%)
443g十二烷基硫酸钠(15%)
223g乙烯基磺酸钠(去离子水中30%)
32g乙酸钠(无水)
5.1g偏亚硫酸氢钠
0.03g的Mohr氏盐(Fe2+盐)
用氮气吹扫反应器去除氧气。从11856g乙酸乙烯酯和52g乙烯基三甲氧基硅烷组成的总量11908g单体混合物中将29%的混合物加入到反应器中的水相。打开乙烯阀,用149g乙烯(全部乙烯的13%)将反应器增压,然后再次关闭(乙烯总量:1144g)。
使反应器温度上升至40℃(起始温度)。在该温度下,将引发剂进料,过二硫酸钠(715g去离子水中40g)以大约215g/h的速率经210分钟加入(聚合反应阶段开始)。根据升温可以清楚地看到聚合开始,该升温在43min内持续至65℃。达到65℃,在若干个阶段中开始添加剩余的单体混合物:1.在20min内添加3.5%的单体混合物(1250g/h),2.将20%(2380g/h)与剩余乙烯(995g乙烯,87%)一起经62min添加(达到大约48bar的最大压力),3.完成乙烯添加之后,在57min内进一步添加28.5%的单体混合物。该时期期间,温度从65℃缓慢陡升至75℃。4.随着最终的19%的单体混合物,然后在28min内使温度从75℃进一步升高至90℃,因此伴随添加结束(单体混合物和引发剂),达到最终温度(在聚合反应阶段的结尾)。将反应器在90℃保持另外60min,然后冷却至大致40℃,排出批料。
实施例D-3-VAE-基共聚物分散体制备
在装有锚式搅拌器(以150rpm运转),加热夹套,计量泵并具有30升容积的压力反应器中添加以下组分的水基溶液:
10322g水(去离子)
173g,去离子水中的聚乙烯醇溶液(15%),即部分水解[88水解度(mol%)],在20℃形成4.50cP±0.50的4%溶液粘度。
557g的C11烷基聚乙二醇醚(28mol环氧乙烷)-非离子型乳化剂(去离子水中70%)
433g十二烷基硫酸钠(15%)
223g乙烯基磺酸钠(去离子水中30%)。
32g乙酸钠(无水)
5.1g偏亚硫酸氢钠
0.03g的Mohr氏盐(Fe2+盐)
用氮气吹扫反应器去除氧气。从11856g乙酸乙烯酯和52g乙烯基三甲氧基硅烷组成的总量11908g单体混合物中将32.5%的混合物加入到反应器中的水相。打开乙烯阀,用149g乙烯(全部乙烯的13%)将反应器增压,然后再次关闭(乙烯总量:1144g)。
反应器温度上升至40℃。在该温度下,将引发剂进料,过二硫酸钠(715g去离子水中40g)以大约245g/h的速率经185分钟加入(聚合反应阶段开始)。根据升温可以清楚地看到聚合开始,该升温在35min内持续至65℃。达到65℃,在2个阶段中开始添加剩余的单体混合物:1.在120min内添加48.5%(2380g/h),在同一时间内反应温度从65℃(起始温度)缓慢陡升至75℃。随着单体添加开始,同样添加剩余乙烯(995g乙烯,87%)(60min内,达到大约48bar的最大压力)。2.随着最终的19%的单体混合物,然后在30min内使温度从75℃进一步升高至90℃,因此伴随添加结束(单体混合物和引发剂),达到最终温度(在聚合反应阶段的结尾)。将反应器在90℃保持另外60min,然后冷却至大致40℃,排出批料。
表8:实施例D-1至D-3的工艺参数:
表9:实施例D-1至D-3中获得的VAE共聚物分散体的性能
表10:用于应用测试的油漆配制料
表11:应用参数缎光漆,46%粘结剂(组成参见表10)
因此,与在低温下得到的产物相比,如果使用一种使用升温的方法(始于较低温度和止于较高温度),所得产物同样显示相同或更高的K值。较高的K值通常可以根据较高的分子量来解释,后者对于许多应用(参见以上)而言是有利的。
总之,上述实验数据显示在30-85℃开始,然后连续陡升至60-160℃的聚合方法的有益效果。最终的最高温度越高,工艺过程将越短。根据实施例A-1,常用的单体进料时间可以从3.5h减少到1h。意外地发现与恒定高温下的方法(实施例A-3)相比,K值(分子量的指示)显著更高。另外可以应用相对有效的后加热阶段。
实施例B-2显示如上所述使用陡升步骤的方法,产生120min的单体缓慢添加时间,和比B-1描述的标准方法获得的高约10个单位的K值。
实施例C-1至C-8说明与恒温反应条件下得到的聚合物相比,由陡升方法得到的聚合物将具有类似的或甚至更高的K值。虽然现有试样剂量时间没有变化,但是随时间而升高的反应温度具有间歇时期时间减少的潜力。部分反应热将被消耗用于加热反应混合物,并且在高温下,如果冷却水温度保持恒定,可以去除更多的热。
实施例D1-D3再次显示使用陡升方法在显著减少(-2h)的反应时间下产生可比较的特性。
- 还没有人留言评论。精彩留言会获得点赞!