一种培育抗TYLCV病毒的番茄的方法、载体及其应用与流程
本发明属于植物基因工程领域,具体而言,涉及培育抗TYLCV病毒的番茄的方法、载体及其应用。
背景技术:
番茄(Lycopersicon esculentum Mill.)是大众喜欢的重要蔬菜,但在番茄生长过程中极其容易感染番茄黄化曲叶病毒(Tomato yellow leaf curl virus,TYLCV)。TYLCV属于双生病毒科(Geminiviridae)菜豆金色花叶病毒属(Begomovirus)病毒,病毒基因组是一个2.7-2.8kb的单链环状DNA,有时还伴有卫星病毒。番茄感染病毒后的症状主要表现为叶片上卷、叶缘及叶脉之间黄化,最后会表现出叶片变小、植株矮化等症状。TYLCV在我国及世界番茄产区广泛流行,对番茄的产量和品质影响极大,经常给番茄生产造成严重的损失,甚至造成绝产绝收。TYLCV在自然条件下通过烟粉虱(Bemisia tabaci)传播。烟粉虱繁殖力强,而且寄主广泛,几乎涵盖所有常见蔬菜、多种花卉等农作物,很难将TYLCV限制在一个地点,阻止其传播与扩散。TYLCV早在1930年在以色列发现,目前在世界上的所有番茄产区均有发现报道。因此,培育抗TYLCV的优良品种,是消除TYLCV危害的主要途径。
近年来,通过重组构建人工核酸酶,能定点切开基因组DNA,从而成功实现对植物基因组的定点、定位修饰。最先出现的是锌指核酸酶(Zinc finger nuclease,ZFN),ZFN技术通过锌指蛋白的锌指域识别特定的DNA序列,与II型限制性内切酶FokI的核酸酶活性域重组,构建成人工重组锌指核酸酶。该重组锌指核酸酶通过其锌指域结合特定的基因组DNA位点,然后来自FokI的核酸酶活性域便能切断该位点附近的DNA。
在ZFN不久之后的2012年,出现了类转录激活子样效应因子核酸酶(Transcription activator-like effector nuclease,TALEN技术)。它基于与ZFN类似的原理,利用TALE(Transcription activator-like effector)蛋白的特异识别DNA碱基的功能域与FokI核酸酶的酶切活性域组合成重组蛋白(TALE nucleases,TALEN)。这样,TALE具有特异性识别基因组特定DNA位点的能力,而FokI的核酸酶活性域便能切断该位点附近的DNA。与ZFN不同的是,TALEN更容易人为地改变其识别并结合DNA碱基序列的能力,从而设计出剪切基因组不同DNA位点的重组核酸酶。
到2013年,出现了能剪切基因组DNA的另一技术,即CRISPR/Cas(clustered regularly interspaced short palindromic repeats/CRISPR-associated proteins)。CRISPR/Cas是存在于细菌和古细菌中的一种适应性免疫防御系统,专门用来对抗入侵细菌的噬菌体的外源DNA。CRISPR/Cas系统通过将入侵细菌的噬菌体DNA的片段整合到CRISPR中,并利用相应的CRISPR RNAs(crRNAs)来指导入侵细菌的噬菌体外源DNA同源序列的降解,从而消灭入侵噬菌体。依此原理,开发出用于高等生物基因组DNA剪切的是其中的一种,即CRISPR/Cas9。其功能与上述ZFN技术及TALEN技术相同,但操作更简便,使用更灵活。在2015年,基于CRISPR/Cas的技术出现了新的称之为Cpf1的技术,它与CRISPR/Cas9极为相似,都是通过转录的RNA结合特定DNA,然后对目标DNA进行剪切。
目前仍然需要更好的方法来培育抗TYLCV的优良番茄品种。
技术实现要素:
为了有效培育出抗TYLCV病毒的番茄,本发明提供了基于核酸内切酶降解入侵番茄细胞的TYLCV病毒的方法、载体及其应用。
具体而言,本发明提供了:
(1)一种培育抗TYLCV病毒的番茄的方法,包括:
1)提供包含核酸内切酶系统的编码序列的DNA序列,其中所述核酸内切酶系统包括导向部分和核酸内切酶活性部分,所述核酸内切酶活性部分本身不具有识别特定核酸序列的功能,其在所述导向部分的作用下能够特异性地切断TYLCV病毒的DNA分子;以及
2)通过转基因方法将包含所述核酸内切酶系统的编码序列的DNA插入番茄的基因组中,使所述番茄表达所述核酸内切酶系统,从而使所述番茄获得抗TYLCV病毒的能力。
(2)根据(1)所述的方法,其中所述核酸内切酶系统为重组核酸内切酶,该重组核酸内切酶包括TYLCV病毒的C1蛋白的N末端作为所述导向部分、以及核酸内切酶的切割活性区作为所述核酸内切酶活性部分,其中所述C1蛋白的N末端能够特异性识别并结合TYLCV病毒的DNA的复制起点区域。
(3)根据(2)所述的方法,其中所述C1蛋白的N末端的核苷酸序列如以下所述:
a)如序列表SEQ ID No.2所示;
b)与序列表SEQ ID No.2所示核苷酸序列的DNA具有至少90%的相似性;或者
c)能够与序列表SEQ ID No.2所示核苷酸序列的DNA杂交,并且为TYLCV病毒的DNA的一部分;
其中如以上a)或b)或c)所述的核苷酸序列的DNA能够编码能够特异性识别并结合TYLCV病毒的DNA的复制起点的所述C1蛋白的N末端。
(4)根据(2)所述的方法,其中所述C1蛋白的N末端如以下所述:
d)其氨基酸序列如序列表SEQ ID No.3所示;或者
e)其氨基酸序列在序列表SEQ ID No.3的基础上经过氨基酸残基的添加和/或替换和/或删除,并且所述C1蛋白的N末端能够特异性识别并结合TYLCV病毒的DNA的复制起点。
(5)根据(2)所述的方法,其中所述核酸内切酶的切割活性区为限制性内切酶的切割活性区。
(6)根据(2)所述的方法,其中所述核酸内切酶的切割活性区为Eco31I的切割活性区,并且核苷酸序列如以下所述:
i)如序列表SEQ ID No.4所示;
ii)与序列表SEQ ID No.4所示核苷酸序列的DNA具有至少90%的相似性,并且能够编码具有核酸内切酶活性的Eco31I的切割活性区;或者
iii)能够与序列表SEQ ID No.4所示核苷酸序列的DNA杂交,并且能够编码具有核酸内切酶活性的Eco31I的切割活性区。
(7)根据(6)所述的方法,其中所述Eco31I的切割活性区如以下所述:
iv)其氨基酸序列如序列表SEQ ID No.5所示;或者
v)其氨基酸序列在序列表SEQ ID No.5的基础上经过氨基酸残基的添加和/或替换和/或删除,并且具有所述Eco31I的切割活性区的活性。
(8)根据(2)所述的方法,其中所述核酸内切酶的切割活性区为SMR切割活性区,并且核苷酸序列如以下所述:
I)如序列表SEQ ID No.6所示;
II)与序列表SEQ ID No.6所示核苷酸序列的DNA具有至少90%的相似性,并且能够编码具有核酸内切酶活性的SMR切割活性区;或者
III)能够与序列表SEQ ID No.6所示核苷酸序列的DNA杂交,并且能够编码具有核酸内切酶活性的SMR切割活性区。
(9)根据(8)所述的方法,其中所述SMR切割活性区如以下所述:
IV)其氨基酸序列如序列表SEQ ID No.7所示;或者
V)其氨基酸序列在序列表SEQ ID No.7的基础上经过氨基酸残基的添加和/或替换和/或删除,并且具有所述SMR切割活性区的活性。
(10)根据(2)和(6)所述的方法,其中所述重组核酸内切酶的导向部分为C1蛋白的N末端,所述重组核酸内切酶的切割活性区为Eco31I的切割活性区,并且所述重组核酸内切酶的核苷酸序列如以下所述:
A)如序列表SEQ ID No.8所示;
B)与序列表SEQ ID No.8所示核苷酸序列的DNA具有至少90%的相似性;或者
C)能够与序列表SEQ ID No.8所示核苷酸序列的DNA杂交;
其中如以上A)或B)或C)所述的核苷酸序列的DNA能够编码特异性识别并结合TYLCV病毒的DNA复制起点且具有核酸内切酶活性的重组核酸内切酶。
(11)根据(10)所述的方法,其中所述重组核酸内切酶如以下所述:
D)其氨基酸序列如序列表SEQ ID No.9所示;或者
E)其氨基酸序列在序列表SEQ ID No.9的基础上经过氨基酸残基的添加和/或替换和/或删除,并且所述重组核酸内切酶能够特异性识别并结合TYLCV病毒的DNA复制起点且具有核酸内切酶活性。
(12)根据(2)和(8)所述的方法,其中所述重组核酸内切酶的导向部分为C1蛋白的N末端,所述重组核酸内切酶的切割活性区为SMR切割活性区,并且所述重组核酸内切酶的核苷酸序列如以下所述:
①如序列表SEQ ID No.10所示;
②与序列表SEQ ID No.10所示核苷酸序列的DNA具有至少90%的相似性;或者
③能够与序列表SEQ ID No.10所示核苷酸序列的DNA杂交;
其中如以上①或②或③所述的核苷酸序列的DNA能够编码特异性识别并结合TYLCV病毒的DNA复制起点且具有核酸内切酶活性的重组核酸内切酶。
(13)根据(12)所述的方法,其中所述重组核酸内切酶如以下所述:
④其氨基酸序列如序列表SEQ ID No.11所示;或者
⑤其氨基酸序列在序列表SEQ ID No.11的基础上经过氨基酸残基的添加和/或替换和/或删除,并且所述重组核酸内切酶能够特异性识别并结合TYLCV病毒的DNA复制起点且具有核酸内切酶活性。
(14)根据(1)所述的方法,其中所述核酸内切酶系统为CRISPR/Cas9系统,该CRISPR/Cas9系统包括与TYLVC病毒DNA中的片段互补的gRNA作为所述导向部分、以及CRISPR/Cas9核酸酶作为所述核酸内切酶活性部分。
(15)根据(14)所述的方法,其中所述CRISPR/Cas9系统包括与TYLVC病毒的正链DNA中的片段互补的一种或多种gRNA。
(16)根据(14)所述的方法,其中所述CRISPR/Cas9系统包括与TYLVC病毒的负链DNA中的片段互补的一种或多种gRNA。
(17)根据(14)所述的方法,其中所述CRISPR/Cas9系统包括与TYLVC病毒的正链DNA中的片段互补的一种或多种gRNA、以及与TYLVC病毒的负链DNA中的片段互补的一种或多种gRNA。
(18)一种在(1)-(17)中任一项所述的培育抗TYLCV病毒的番茄的方法中使用的载体。
(19)根据(18)所述的载体在培育抗TYLCV病毒的番茄中的应用。
本发明与现有技术相比具有以下优点和积极效果:
通过本发明的构思和方法,本发明所构建的核酸内切酶系统能够特异性地切断TYLCV病毒的DNA分子,从而本发明成功地获得了具有抗TYLCV病毒的能力的番茄品种。
本发明对于培育抗番茄曲叶病毒病的番茄具有重大价值。
附图说明
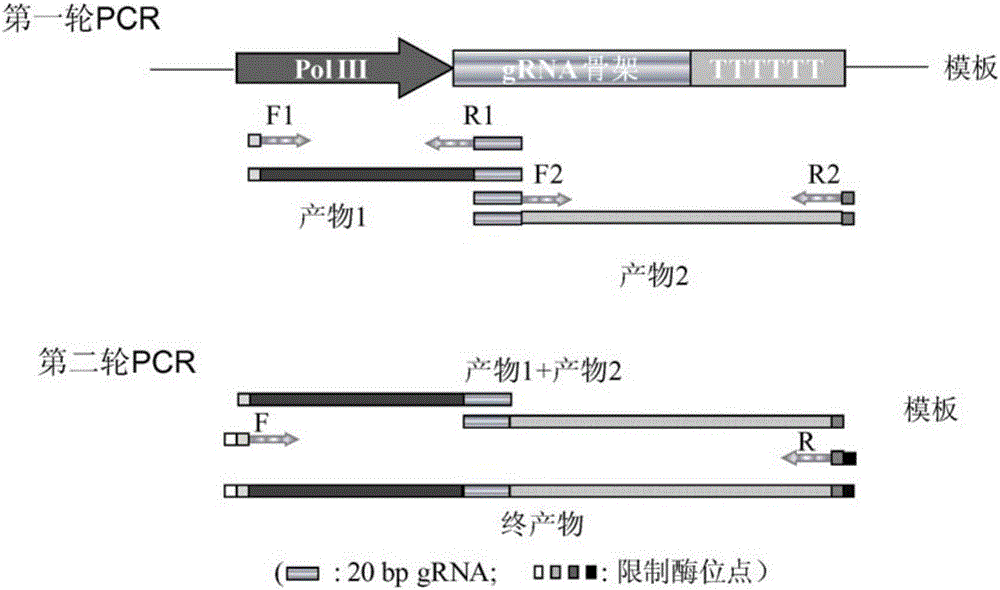
图1示出重叠PCR方法及过程示意图。
图2示出重叠PCR构建gRNA表达盒的方法及过程示意图。
图3示出本发明一个实施方案中通过双酶切法构建pBI121+Cas9-gRNA(F-LIR)载体的示意图。
图4示出本发明一个实施方案中通过双酶切法构建pBI121+Cas9-gRNA(R-LIR)载体的示意图。
图5示出野生型番茄通过农杆菌接种TYLCV病毒后的形态。
图6示出本发明的转基因番茄通过农杆菌接种TYLCV病毒后的形态。
图7示出野生型番茄通过烟粉虱接种TYLCV病毒后的形态。
图8示出本发明的转基因番茄通过烟粉虱接种TYLCV病毒后的形态。
图9示出野生型番茄在田间感染TYLCV病毒后的形态。
图10示出本发明的转基因番茄在田间不受TYLCV病毒感染的形态。
具体实施方式
以下通过具体实施方式的描述并参照附图对本发明作进一步说明,但这并非是对本发明的限制,本领域技术人员根据本发明的基本思想,可以做出各种修改或改进,但是只要不脱离本发明的基本思想,均在本发明的范围之内。
为了开发更好的方法来培育抗TYLCV病毒的优良番茄品种,本发明的发明人进行了认真的理论研究和试验摸索,最终形成了本发明的巧妙构思和有效的方法,即:对侵入细胞的病毒DNA进行降解,从而阻断病毒的复制,有效地控制病毒病的发展。据此,通过转基因方法将编码切断TYLCV病毒DNA分子的核酸内切酶系统的DNA转入番茄,使番茄表达该核酸内切酶系统,从而有效阻止TYLCV病毒的DNA复制,抑制TYLCV病毒在番茄体内的传播,由此提供抗TYLCV病毒的优良番茄品种。
具体而言,本发明提供了一种培育抗TYLCV病毒的番茄的方法,包括:
1)提供包含核酸内切酶系统的编码序列的DNA序列,其中所述核酸内切酶系统包括导向部分和核酸内切酶活性部分,所述核酸内切酶活性部分本身不具有识别特定核酸序列的功能,其在所述导向部分的作用下能够特异性地切断TYLCV病毒的DNA分子;以及
2)通过转基因方法将包含所述核酸内切酶系统的编码序列的DNA插入番茄的基因组中,使所述番茄表达所述核酸内切酶系统,从而使所述番茄获得抗TYLCV病毒的能力。
在本发明中,术语“核酸内切酶系统”是指具有核酸内切酶活性的复合体,其包括导向部分和核酸内切酶活性部分。核酸内切酶系统可以是重组蛋白,也可以是包括其他具有导向作用的分子(例如RNA)和具有核酸内切酶活性的蛋白的复合体。
本文所用术语“导向部分”是指核酸内切酶系统中能够引导核酸内切酶活性部分定点切割目标核酸序列的部分,其可以是例如多肽、蛋白、RNA等。
本文所用术语“核酸内切酶活性部分”是指核酸内切酶系统中具有核酸内切酶活性,但本身不具有识别特定核酸序列的特异性功能的部分,需要在所述“导向部分”的引导下在特定的位点切割DNA。该核酸内切酶活性部分可以是完整的核酸内切酶(例如CRISPR/Cas9核酸酶),也可以是分别具有独立的核酸识别区域和核酸切割区域的核酸内切酶的核酸切割区域或其一部分(只要能够发挥核酸内切酶活性即可)。
在本发明的一个实施方案中,所述核酸内切酶系统为重组核酸内切酶,该重组核酸内切酶包括TYLCV病毒的C1蛋白的N末端作为所述导向部分、以及核酸内切酶的切割活性区作为所述核酸内切酶活性部分,其中所述C1蛋白的N末端能够特异性识别并结合TYLCV病毒的DNA的复制起点区域。
所述术语“核酸内切酶的切割活性区”即是指分别具有独立的核酸识别区域和核酸切割区域的核酸内切酶的核酸切割区域或其中一部分(只要能够发挥核酸内切酶活性即可)。
在TYLCV基因组中,C1基因编码一种负责病毒DNA复制的蛋白(即C1蛋白,GenBank:AGG35491.1),该蛋白特异性地结合病毒DNA,并借助寄主细胞的复制体系,启动病毒基因组的复制。C1蛋白的N-末端包含能特异性识别病毒的复制起点、并与复制起点相结合的区域;而C-末端有一区域与病毒DNA复制的启动相关。据此,本发明构建了具有能够特异性识别并结合病毒复制起点区域的C1蛋白N-末端和核酸内切酶的切割活性区的重组核酸酶。通过将该重组核酸酶的DNA片段克隆到植物表达载体中,使其在番茄体内表达,从而在入侵病毒的复制起点区域切断病毒DNA分子,进而有效阻断病毒的复制、扩散。所述C1蛋白N-末端所识别和结合的病毒复制起点区域如文献“Campos-Olivas,R.,Louis,J.M.,Clérot,D.,Gronenborn,B.,&Gronenborn,A.M.2002.The structure of a replication initiator unites diverse aspects of nucleic acid metabolism.Proceedings of the National Academy of Sciences,99:10310-10315”所述。
编码C1蛋白的N-末端DNA片段可以采用PCR的方法从TYLCV病毒的基因组(基因组序列如序列表SEQ ID No.1所示,GenBank:KC211184.1)中克隆得到,所用引物序列依克隆方法及所重组连接的核酸内切酶的切割活性区的不同有所差异。本领域技术人员理解克隆获得C1蛋白的N-末端可以采用本领域已知的常规方法进行。
编码核酸内切酶的切割活性区DNA片段同样可以采用常规的PCR方法克隆得到。
重组核酸内切酶的DNA分子可以通过重组PCR方法和/或限制性内切酶的方法克隆到植物表达载体中。这些方法和所用材料都可采用本领域已知的常规方法和材料。
在本发明一个具体的实施方案中,所述C1蛋白的N末端的核苷酸序列如以下所述:
a)如序列表SEQ ID No.2所示;
b)与序列表SEQ ID No.2所示核苷酸序列的DNA具有至少90%(例如91%、92%、93%、94%、95%、96%、97%、98%、或99%)的相似性;或者
c)能够与序列表SEQ ID No.2所示核苷酸序列的DNA杂交,并且为TYLCV病毒的DNA的一部分;
其中如以上a)或b)或c)所述的核苷酸序列的DNA能够编码能够特异性识别并结合TYLCV病毒的DNA的复制起点的所述C1蛋白的N末端。
优选地,所述C1蛋白的N末端如以下所述:
d)其氨基酸序列如序列表SEQ ID No.3所示;或者
e)其氨基酸序列在序列表SEQ ID No.3的基础上经过氨基酸残基的添加和/或替换和/或删除,并且所述C1蛋白的N末端能够特异性识别并结合TYLCV病毒的DNA的复制起点。
在一个实施方案中,在本发明所述的重组核酸内切酶中,所述核酸内切酶的切割活性区为限制性内切酶的切割活性区。只要给定能够引导核酸内切酶活性部分定点切割TYLCV病毒DNA的导向部分,那么其所重组连接的限制性内切酶的切割活性区则可以来自任何分别具有独立的核酸识别区域和核酸切割区域的限制性内切酶。
优选地,所述核酸内切酶为Eco31I(GenBank:AAM09638.2)或SMR(small MutS-related domain,GenBank:AAZ37220.1),所述核酸内切酶的切割活性区为Eco31I的切割活性区或SMR切割活性区。
在Eco31I的切割活性区的情况中,Eco31I的切割活性区可以为Eco31I的第250位氨基酸至第579位氨基酸。
优选地,Eco31I的切割活性区的核苷酸序列如以下所述:
i)如序列表SEQ ID No.4所示;
ii)与序列表SEQ ID No.4所示核苷酸序列的DNA具有至少90%(例如91%、92%、93%、94%、95%、96%、97%、98%、或99%)的相似性,并且能够编码具有核酸内切酶活性的Eco31I的切割活性区;或者
iii)能够与序列表SEQ ID No.4所示核苷酸序列的DNA杂交,并且能够编码具有核酸内切酶活性的Eco31I的切割活性区。
优选地,所述Eco31I的切割活性区如以下所述:
iv)其氨基酸序列如序列表SEQ ID No.5所示;或者
v)其氨基酸序列在序列表SEQ ID No.5的基础上经过氨基酸残基的添加和/或替换和/或删除,并且所述Eco31I的切割活性区具有核酸内切酶活性。
在Eco31I的切割活性区的情况中,所述重组核酸内切酶的核苷酸序列优选如以下所述:
A)如序列表SEQ ID No.8所示;
B)与序列表SEQ ID No.8所示核苷酸序列的DNA具有至少90%(例如91%、92%、93%、94%、95%、96%、97%、98%、或99%)的相似性;或者
C)能够与序列表SEQ ID No.8所示核苷酸序列的DNA杂交;
其中如以上A)或B)或C)所述的核苷酸序列的DNA能够编码特异性识别并结合TYLCV病毒的DNA复制起点且具有核酸内切酶活性的重组核酸内切酶。
优选地,所述重组核酸内切酶如以下所述:
D)其氨基酸序列如序列表SEQ ID No.9所示;或者
E)其氨基酸序列在序列表SEQ ID No.9的基础上经过氨基酸残基的添加和/或替换和/或删除,并且所述重组核酸内切酶能够特异性识别并结合TYLCV病毒的DNA复制起点且具有核酸内切酶活性。
在SMR切割活性区的情况中,SMR切割活性区可以为假单胞菌一SMR功能蛋白的第95位氨基酸至第185位氨基酸。
优选地,SMR切割活性区的核苷酸序列如以下所述:
I)如序列表SEQ ID No.6所示;
II)与序列表SEQ ID No.6所示核苷酸序的DNA列具有至少90%(例如91%、92%、93%、94%、95%、96%、97%、98%、或99%)的相似性,并且能够编码具有核酸内切酶活性的SMR切割活性区;或者
III)能够与序列表SEQ ID No.6所示核苷酸序列的DNA杂交,并且能够编码具有核酸内切酶活性的SMR切割活性区。
优选地,所述SMR切割活性区如以下所述:
IV)其氨基酸序列如序列表SEQ ID No.7所示;或者
V)其氨基酸序列在序列表SEQ ID No.7的基础上经过氨基酸残基的添加和/或替换和/或删除,并且所述SMR切割活性区具有核酸内切酶活性。
在SMR切割活性区的情况中,所述重组核酸内切酶的核苷酸序列优选如以下所述:
①如序列表SEQ ID No.10所示;
②与序列表SEQ ID No.10所示核苷酸序列的DNA具有至少90%(例如91%、92%、93%、94%、95%、96%、97%、98%、或99%)的相似性;或者
③能够与序列表SEQ ID No.10所示核苷酸序列的DNA杂交;
其中如以上①或②或③所述的核苷酸序列的DNA能够编码特异性识别并结合TYLCV病毒的DNA复制起点且具有核酸内切酶活性的重组核酸内切酶。
优选地,所述重组核酸内切酶如以下所述:
④其氨基酸序列如序列表SEQ ID No.11所示;或者
⑤其氨基酸序列在序列表SEQ ID No.11的基础上经过氨基酸残基的添加和/或替换和/或删除,并且所述重组核酸内切酶能够特异性识别并结合TYLCV病毒的DNA复制起点且具有核酸内切酶活性。
在本发明的另一个实施方案中,为了控制病毒DNA复制,本发明还构建了CRISPR/Cas9系统,该CRISPR/Cas9系统包括与TYLVC病毒DNA中的片段互补的gRNA作为所述导向部分、以及CRISPR/Cas9核酸酶作为所述核酸内切酶活性部分。
所述gRNA与互补的病毒DNA片段配对形成DNA:RNA杂交分子,从而引导CRISPR/Cas9核酸酶与DNA:RNA杂交分子结合,由此特异性地切断DNA:RNA杂交位点附近的病毒DNA,达到有效阻断病毒DNA复制的效果,从而让番茄获得抗番茄黄化曲叶病毒病的能力。
已知CRISPR/Cas9的酶切活性依赖杂交DNA旁边的PAM序列,即NGG(N代表A、T、C和G四个碱基中的任何一个碱基)。因此,在TYLCV基因组中只要有NGG,就可以在其附近以病毒DNA为模板设计引导RNA,即gRNA。模板可以包括病毒DNA的病毒链(正链)及互补链(负链)。
在本发明中,所述CRISPR/Cas9系统可以包括与TYLVC病毒的正链DNA中的片段互补的一种或多种gRNA;也可以包括与TYLVC病毒的负链DNA中的片段互补的一种或多种gRNA;还可以包括与TYLVC病毒的正链DNA中的片段互补的一种或多种gRNA、和与TYLVC病毒的负链DNA中的片段互补的一种或多种gRNA。也即,引导CRISPR/Cas9结合TYLCV基因组DNA的gRNA编码序列在植物表达载体中可以有一个,也可以有多个,而且,gRNA可以与病毒DNA的病毒链特异结合,也可与病毒DNA的互补链配对。
在确定gRNA的目标序列之后,可以通过分子生物学领域常规的克隆方法获得编码gRNA的DNA序列。
本发明使用的CRISPR/Cas9核酸酶的编码序列是已知的(可参见文献“Li,J.F.,Norville,J.E.,Aach,J.,McCormack,M.,Zhang,D.,Bush,J.,Church G.M.,and Sheen,J.2013.Multiplex and homologous recombination-mediated genome editing in Arabidopsis and Nicotiana benthamiana using guide RNA and Cas9.Nature biotechnology,31:688-691”)。可以将CRISPR/Cas9和所设计的编码gRNA的DNA克隆到植物表达载体,其方法可以采用分子生物学领域常规的方法,例如图1、图2和3所示的方法。
在克隆得到含有本发明所述编码核酸内切酶系统的DNA(例如载体)之后,将所述DNA插入番茄的基因组中,使所述番茄表达所述核酸内切酶系统,从而使所述番茄获得抗TYLCV病毒的能力。
通常,可以将含有本发明所述编码核酸内切酶系统的DNA克隆到植物表达载体中,在转入番茄之后让番茄表达所述核酸内切酶系统。所述植物表达载体可以采用本领域通常所用的任何植物表达载体。
将含有本发明所述编码核酸内切酶系统的DNA克隆到植物表达载体中,并将该载体转入番茄的方法是本领域已知的,并且可以采用本领域常规所用的材料和试剂,其过程可以包括,例如,农杆菌感受态细胞的制备、用载体质粒转化农杆菌、用经转化的农杆菌转化番茄无菌苗子叶或下胚轴外植体、外源DNA插入和表达的鉴定、抗TYLCV病毒的鉴定等。
本发明还提供了一种在本发明中所述的培育抗TYLCV病毒的番茄的方法中使用的载体。
所述载体包含含有本发明所述核酸内切酶系统的编码序列的DNA,其中所述核酸内切酶系统包括导向部分和核酸内切酶活性部分,所述核酸内切酶活性部分本身不具有识别特定核酸序列的功能,其在所述导向部分的作用下能够特异性地切断TYLCV病毒的DNA分子。
所述载体可以为本领域已知的任何常用的载体,例如包括克隆载体、植物表达载体等。
本发明还提供了所述载体在培育抗TYLCV病毒的番茄中的应用。
本发明所公开的构思和方法还可应用于在其它植物中获得抗双生病毒等DNA病毒感染的能力。“双生病毒”是指来自植物病毒双生病毒科(Geminiviridae)的病毒。双生病毒可包括但不限于大麦黄矮病毒、棉花缩叶病毒、番茄黄化曲叶病毒、辣椒曲叶病毒、豆金色花叶病毒、豆金色花叶病毒、菜豆矮花叶病毒、茼麻花叶病毒、非洲木薯花叶病毒、烟草曲叶病毒和南瓜曲叶病毒等等。
以下通过例子的方式进一步解释或说明本发明的内容,但这些例子不应被理解为对本发明的保护范围的限制。
实施例
下述实施例中的实验方法,除有特殊说明外,均为常规方法。下述实施例中所用的实验材料,如无特殊说明,均采购自市场出售的生化试剂。
实施例1:构建重组核酸酶C1-E表达载体
第一步,根据病毒C1基因的序列,设计如下PCR引物,以病毒C1基因为模板扩增识别TYLCV复制起点区域的C1蛋白N末端的DNA片段。
F1(C1-N-F):
5’-GGATCCATGCCTCGTTTATTTAAAATATATGCC-3’,(下划线示出BamHI酶切位点)
R1(OL-AC1/ECO31I-R):
5’-TTAAGATTTTCTTTGGTCCTACCTCCTGGCCGCGCAGCGG-3’
第二步,用如下引物,以编码Eco31I的基因序列为模板扩增Eco31I的切割活性区的DNA片段。
F2(OL-AC1/ECO31I-F):
5’-CCGCTGCGCGGCCAGGAGGTAGGACCAAAGAAAATCTTAA-3’
R2(Eco31I-R):
5’-TCTAGATTACTCTATAAATTCTTCTGGGAT-3’,(下划线示出XbaI酶切位点)
第三步,用上述第一步和第二步扩增得到的DNA片段混合作为模板,利用重叠PCR(引物:F1(C1-N-F)和R2(Eco31I-R);重叠PCR方法如图1所示)扩增得到C1蛋白N末端编码DNA与Eco31I切割活性区编码DNA的重组基因C1-E,如序列表SEQ ID No.8。C1-E经TA克隆的方法连接到pMD18-T载体(购自TAKARA,货号D101A),并经DNA测序验证。
第四步,利用双酶切法将重组基因C1-E克隆到植物表达载体pHB(参见文献“Mao,J.,Zhang,Y.C.,Sang,Y.,Li,Q.H.,&Yang,H.Q.2005.A role for Arabidopsis cryptochromes and COP1in the regulation of stomatal opening.Proceedings of the National Academy of Sciences of the United States of America,102:12270-12275”)的BamHI和XbaI位点中,得到重组核酸酶C1-E表达载体。
实施例2:构建重组核酸酶C1-S表达载体
第一步,根据病毒基因C1序列,设计如下PCR引物,以病毒C1基因为模板扩增识别TYLCV复制起点区域的C1蛋白N末端的DNA片段。
F1(C1-N-F):
5’-GGATCCATGCCTCGTTTATTTAAAATATATGCC-3’,(下划线示出BamHI酶切位点)
R1(OL-AC1/SMR-R):
5’-CTCATGCCGTGTAAGTCCAGACCGCCACCTCCTGGCCGCG-3’
第二步,用如下引物,以SMR的编码序列为模板扩增SMR切割活性区的DNA片段。
F2(OL-AC1/SMR-F):
5’-CGCGGCCAGGAGGTGGCGGTCTGGACTTACACGGCATGAG-3’
R2(SMR-R):5’-TCTAGATTACTCGTCGCGTCCTTCC-3’,(下划线示出XbaI酶切位点)
第三步,用上述第一步和第二步扩增得到的DNA片段混合作模板,利用重叠PCR(引物:F1(C1-N-F)和R2(SMR-R);重叠PCR方法如图1所示)扩增得到C1蛋白N末端与SMR切割活性区的重组基因C1-S,如序列表SEQ ID No.10。C1-S经TA克隆的方法连接到pMD18-T载体(购自TAKARA,货号D101A),并经DNA测序验证。
第四步,利用双酶切法将重组基因C1-S克隆到植物表达载体pHB的BamHI和XbaI位点中,得到重组核酸酶C1-S表达载体。
实施例3:构建具有一个与TYLVC病毒正链DNA互补的gRNA的CRISPR/Cas9表达载体
1、选定TYLVC病毒正链的大间隔区(large intergenic region,LIR)的如下序列作为CRISPR/Cas9系统中的gRNA目标序列:
5’-ATCCGTATAATATTACCGGATGG-3’
2、U6::gRNA(F-LIR)表达盒的PCR扩增及克隆
采用重叠PCR技术扩增U6-gRNA(F-LIR)的DNA片段,重叠PCR扩增方法如图1所示。跟据TYLCV正链的大间隔区DNA序列确定的gRNA目标序列及HBT-pcoCas9质粒(http://www.addgene.org/,质粒编号:52254)设计引物:F1、R2、R1和F2,第一轮扩增模板为HBT-pcoCas9质粒DNA。
F1:5’-GGGCCCAGAAATCTCAAAATTCCGGC-3’,(下划线示出ApaI酶切位点)
R2:
5’-GTCGACTAATGCCAACTTTGTACAAGAAAG-3’,(下划线示出SalI酶切位点,双下划线示出EcoRI酶切位点)
R1:
5’-TCCGGTAATATTATACGGATAATCACTACTTCGTCTCT-3’
F2:
5’-ATCCGTATAATATTACCGGAGTTTTAGAGCTAGAAATAGC-3’
重叠PCR扩增DNA片段经TA克隆法连接到pMD18-T载体(购自TAKARA,货号D101A),克隆得到的U6-gRNA(F-LIR)片段均经DNA测序验证。
3、SPA终止子(SPA-Ter)的PCR扩增及克隆
扩增所用引物:
SPAter-F:5’-CTGCAGGTAAGTCATTGAAAAATTTGC-3’,(下划线示出PstI酶切位点);
SPAter-R:5’-AAGCTTCTTTGTTAACCATCATTTAGTG-3’,(下划线示出HindIII酶切位点)。
以大豆基因组DNA为模板扩增出SPA终止子(Soybean-derived poly(A)signal,SPA-Ter)DNA序列(序列表SEQ ID No.12)。
得到的PCR产物经TA克隆法连接到pMD18-T(购自TAKARA,货号D101A)载体,克隆的终止子DNA片段经测序验证。
4、表达gRNA的CRISPR/Cas9载体的构建
用双酶切(SalI和ApaI)法将上述U6-gRNA(F-LIR)片段从pMD18-T载体上酶切下来,克隆到载体pGreenII 0179(http://www.pgreen.ac.uk/)上,同时利用双酶切(BamHI和PstI)法将pcoCas9基因从HBT-pcoCas9质粒切下,连接到载体pGreenII 0179上,之后再用双酶切(PstI和HindIII)法将终止子(SPA-Ter)也连接到载体pGreenII 0179上,最终得到pGreenII 0179+pcoCas9-gRNA(F-LIR)载体。U6::gRNA(F-LIR)表达盒、pcoCas9基因、终止子(SPA-Ter)在载体pGreenII 0179上的连接顺序参见图3。
为了便于对番茄进行转化及筛选转化植株,对载体pGreenII0179-pcoCas9+gRNA(F-LIR)进行双酶切(BamHI和EcoRI),将得到的pcoCas9+gRNA(F-LIR)片段连接到植物表达载体pBI121(https://www.arabidopsis.org/,质粒编号:3CD3-388)上,最后得到植物表达载体pBI121+pcoCas9-gRNA(F-LIR)(见图3)。
实施例4:构建具有一个与TYLVC病毒负链DNA互补的gRNA的CRISPR/Cas9表达载体
1、选定TYLVC病毒负链的大间隔区(large intergenic region,LIR)的如下序列作为CRISPR/Cas9系统中的gRNA目标序列:
5’-ATCCGGTAATATTATACGGATGG-3’
2、U6::gRNA(R-LIR)表达盒的PCR扩增及克隆
采用重叠PCR技术扩增U6-gRNA(R-LIR)的DNA片段,重叠PCR扩增方法如图1所示。跟据TYLCV负链的大间隔区DNA序列确定的gRNA目标序列及HBT-pcoCas9质粒(http://www.addgene.org,质粒编号:52254),设计引物:F1、R2、R1和F2:第一轮扩增模板为HBT-pcoCas9质粒DNA。
F1:5’-GGGCCCAGAAATCTCAAAATTCCGGC-3’,(下划线示出ApaI酶切位点)
R2:5’-GTCGACTAATGCCAACTTTGTACAAGAAAG-3’,(下划线示出SalI酶切位点,双下划线示出EcoRI酶切位点)
R1:
5’-TCCGTATAATATTACCGGATAATCACTACTTCGTCTCT-3’
F2:
5’-ATCCGGTAATATTATACGGAGTTTTAGAGCTAGAAATAGC-3’
重叠PCR扩增DNA片段经TA克隆法连接到pMD18-T(购自TAKARA,货号D101A)载体,得到的克隆U6-gRNA(R-LIR)片段经DNA测序验证。
3、SPA终止子(SPA-Ter)的PCR扩增
扩增所用引物:
SPAter-F:5’-CTGCAGGTAAGTCATTGAAAAATTTGC-3,(下划线示出PstI酶切位点);
SPAter-R:5’-AAGCTTCTTTGTTAACCATCATTTAGTG-3’,(下划线示出HindIII酶切位点)。
以大豆基因组DNA为模板扩增出SPA终止子(Soybean-derived poly(A)signal,SPA-Ter)的DNA序列(序列表SEQ ID No.12)。
扩增得到的PCR产物经TA克隆法,连接到pMD18-T(购自TAKARA,货号D101A)载体,克隆的终止子DNA片段经测序验证。
4、表达gRNA的CRISPR/Cas9载体的构建
用双酶切(SalI和ApaI)法将上述U6-gRNA(R-LIR)片段从pMD18-T载体上酶切下来,然后插入到载体pGreenII 0179(http://www.pgreen.ac.uk/)上,同时利用双酶切(BamHI和PstI)法将pcoCas9基因从HBT-pcoCas9质粒切下,连接到载体pGreenII 0179上,之后再用双酶切(PstI和HindIII)法将终止子(SPA-Ter)也连接到载体pGreenII 0179上,最终形成pGreenII 0179+pcoCas9-gRNA(R-LIR)载体。U6::gRNA(R-LIR)表达盒、pcoCas9基因、终止子(SPA-Ter)在载体pGreenII 0179上的连接顺序参见图4。
为了便于对番茄进行转化及筛选转化植株,对载体pGreenII 0179+pcoCas9-gRNA(R-LIR)进行双酶切(BamHI和EcoRI),将得到的pcoCas9-gRNA(R-LIR)片段连接到植物表达载体pBI121(https://www.arabidopsis.org/,质粒编号:3CD3-388)上,最后得到植物表达载体pBI121+pcoCas9-gRNA(R-LIR)(见图4)。
实施例5:构建具有4个与TYLVC病毒负链DNA互补的gRNA的CRISPR/Cas9表达载体
1、对于CRISPR/Cas9系统中的gRNA,选定TYLVC病毒的目标序列如下:
1)F1:5’-CATTTCCACGCCCGTCTCGAAGG-3’,位于TYLCV基因组的V1基因;
2)F2:5’-GCTGCTGTCCCCATTGTCCAAGG-3’,位于TYLCV基因组的V1基因;
3)F3:5’-TCTTCTTGGTTCGTGATAGAAGG-3’,位于TYLCV基因组的V1基因;
4)F4:ATCCGTATAATATTACCGGATGG-3’,位于TYLCV基因组的大间隔区(large intergenic region,LIR)。
2、CRISPR/Cas9载体中的4个gRNA表达盒的PCR扩增及克隆
每个gRNA表达盒均采用重叠PCR方法扩增并克隆到T载体,重叠PCR扩增方法如图2所示。
1)gRNA表达盒1(目标序列F1):AtU3b::gRNA-F1
所设计的PCR引物:
F1(U-F):5’-CTCCGTTTTACCTGTGGAATCG-3’;
R2(gR-R):5’-CGGAGGAAAATTCCATCCAC-3’;
R1(AtU3b-F1):
5’-TCGAGACGGGCGTGGAAATGTGACCAATGTTGCTCC-3’;
F2(gRTF1):
5’-CATTTCCACGCCCGTCTCGAGTTTTAGAGCTAGAAAT-3’;
F(Pps-GGL-1):
5’-TTCAGAGGTCTCTCTCGATGGAATCGGCAGCAAAGG-3’,(下划线示出BsaI酶切位点,双下划线示出SpeI酶切位点);
R(Pps-GG2-1):
5’-AGCGTGGGTCTCGTCAGGGTCCATCCACTCCAAGCTC-3’,(下划线示出BsaI酶切位点)。
以pYLgRNA-AtU3b(GenBank:KR029097)质粒为模板进行第一轮扩增(所用引物:F1(U-F)、R2(gR-R)、R1(AtU3b-F1)、F2(gRTF1)),第二轮扩增用引物F(Pps-GGL-1)和R(Pps-GG2-1)。扩增片段直接连接到ZT4-blunt载体(购自庄盟生物,货号ZC205),并经DNA测序验证。得到的质粒经BsaI酶切分离片段:
2)gRNA表达盒2(目标序列F2):AtU3d::gRNA-F2
所设计的PCR引物:
F1(U-F):5’-CTCCGTTTTACCTGTGGAATCG-3’;
R2(gR-R):5’-CGGAGGAAAATTCCATCCAC-3’;
R1(AtU3d-F2):
5’-TGGACAATGGGGACAGCAGCTGACCAATGGTGCTTTG-3’;
F2(gRTF2):
5’-GCTGCTGTCCCCATTGTCCAGTTTTAGAGCTAGAAAT-3’;
F(Pps-GG2-2):
5’-TTCAGAGGTCTCTCTGACACTGGAATCGGCAGCAAAGG-3’,(下划线示出BsaI酶切位点);
R(Pps-GG3-1):
5’-AGCGTGGGTCTCGTCTTCACTCCATCCACTCCAAGCTC-3’,(下划线示出BsaI酶切位点)。
以pYLgRNA-AtU3d(GenBank:KR029099)质粒为模板进行第一轮扩增(所用引物:F1(U-F)、R2(gR-R)、R1(AtU3d-F2)、F2(gRTF2)),第二轮扩增用引物F(Pps-GG2-2)和R(Pps-GG3-1)。扩增片段直接连接到ZT4-blunt载体(购自庄盟生物,货号ZC205)载体,并经DNA测序验证。得到的质粒经BsaI酶切分离片段:
3)gRNA表达盒3(目标序列F3):AtU6-1::gRNA-F3
所设计的PCR引物:
F1(U-F):5’-CTCCGTTTTACCTGTGGAATCG-3’;
R2(gR-R):5’-CGGAGGAAAATTCCATCCAC-3’;
R1(AtU6-1-F3):
5’-TCTATCACGAACCAAGAAGACAATCACTACTTCGTCT-3’;
F2(gRTF3):
5’-TCTTCTTGGTTCGTGATAGAGTTTTAGAGCTAGAAAT-3’;
F(Pps-GG3-2):
5’-TTCAGAGGTCTCTAAGACTTTGGAATCGGCAGCAAAGG-3’,(下划线示出BsaI酶切位点);
R(Pgs-GG4):
5’-AGCGTGGGTCTCGAGTCCTTTCCATCCACTCCAAGCTC-3’,(下划线示出BsaI酶切位点)。
以pYLgRNA-AtU6-1(GenBank:KR029101)质粒为模板进行第一轮扩增(所用引物:F1(U-F)、R2(gR-R)、R1(AtU6-1-F3)、F2(gRTF3)),第二轮扩增用引物F(Pps-GG3-2)和R(Pgs-GG4)。扩增片段直接连接到ZT4-blunt载体(购自庄盟生物,货号ZC205)载体,并经DNA测序验证。得到的质粒经BsaI酶切分离片段:
4)gRNA表达盒4(目标序列F4):AtU6-29::gRNA-F4
所设计的PCR引物:
F1(U-F):5’-CTCCGTTTTACCTGTGGAATCG-3’;
R2(gR-R):5’-CGGAGGAAAATTCCATCCAC-3’;
R1(AtU6-29-F4):
5’-TCCGGTAATATTATACGGATCAATCTCTTAGTCGACT-3’;
F2(gRTF4):
5’-ATCCGTATAATATTACCGGAGTTTTAGAGCTAGAAAT-3’;
F(Pps-GG4):
5’-TTCAGAGGTCTCTGACTACATGGAATCGGCAGCAAAGG-3’,(下划线示出BsaI酶切位点);
R(Pgs-GGR):
5’-AGCGTGGGTCTCGACCGATCCATCCACTCCAAGCTC-3’,(下划线示出BsaI酶切位点,粗下划线示出MluI酶切位点,双下划线示出SpeI酶切位点)。
以pYLgRNA-AtU6-29(GenBank:KR029102)质粒为模板进行第一轮扩增(所用引物:F1(U-F)、R2(gR-R)、R1(AtU6-29-F4)、F2(gRTF4)),第二轮扩增用引物F(Pps-GG4)和R(Pgs-GGR)。扩增片段直接连接到ZT4-blunt载体(购自庄盟生物,货号ZC205)载体,并经DNA测序验证。得到的质粒经BsaI酶切分离片段:
3、将上述得到的4种经BsaI酶切分离的片段连接到载体pYLCRISPR/cas9P35S-N(GenBank:KR029112)中,其中4种gRNA表达盒的连接顺序(5’至3’方向)如下:
实施例6:构建具有4个与TYLVC病毒正链DNA互补的gRNA的CRISPR/Cas9表达载体
1、对于CRISPR/Cas9系统中的gRNA,选定TYLVC病毒的目标序列如下:
1)R1:5’-CTTCGGCGAACCTTCGAGACGGG-3’,位于TYLCV基因组的V1基因;
2)R2:5’-GGACAATGGGGACAGCAGCACGG-3’,位于TYLCV基因组的V1基因;
3)R3:5’-TTCTTCACGGTTGCGGTACTGGG-3’,位于TYLCV基因组的V1基因;
4)R4:5’-ATCCGGTAATATTATACGGATGG-3’,位于TYLCV基因组的大间隔区(large intergenic region,LIR)。
2、CRISPR/Cas9载体中的各gRNA表达盒的PCR扩增及克隆
每个gRNA表达盒均采用重叠PCR方法扩增并克隆到T载体,重叠PCR扩增方法如图2所示进行。
1)gRNA表达盒1(目标序列R1):AtU3b::gRNA-R1
所设计的PCR引物:
F1(U-F):5’-CTCCGTTTTACCTGTGGAATCG-3’;
R2(gR-R):5’-CGGAGGAAAATTCCATCCAC-3’;
R1(AtU3b-R1):
5’-GTCTCGAAGGTTCGCCGAAGTGACCAATGTTGCTCC-3’;
F2(gRTR1):
5’-CTTCGGCGAACCTTCGAGACGTTTTAGAGCTAGAAAT-3’;
F(Pps-GGL-1):
5’-TTCAGAGGTCTCTCTCGATGGAATCGGCAGCAAAGG-3’,(下划线示出BsaI酶切位点,双下划线示出SpeI酶切位点);
R(Pps-GG2-1):
5’-AGCGTGGGTCTCGTCAGGGTCCATCCACTCCAAGCTC-3’,(下划线示出BsaI酶切位点)。
以pYLgRNA-AtU3b(GenBank:KR029097)质粒为模板进行第一轮扩增(所用引物:F1(U-F)、R2(gR-R)、R1(AtU3b-R1)、F2(gRTR1)),第二轮扩增用引物F(Pps-GGL-1)和R(Pps-GG2-1)。扩增片段直接连接到ZT4-blunt载体(购自庄盟生物,货号ZC205)载体,并经DNA测序验证。得到的质粒经BsaI酶切分离片段:
2)gRNA表达盒2(目标序列R2):AtU3d::gRNA-R2
所设计的PCR引物:
F1(U-F):5’-CTCCGTTTTACCTGTGGAATCG-3’;
R2(gR-R):5’-CGGAGGAAAATTCCATCCAC-3’;
R1(AtU3d-R2):
5’-TGCTGCTGTCCCCATTGTCCTGACCAATGGTGCTTTG-3’;
F2(gRTR2):
5’-GGACAATGGGGACAGCAGCAGTTTTAGAGCTAGAAAT-3’;
F(Pps-GG2-2):
5’-TTCAGAGGTCTCTCTGACACTGGAATCGGCAGCAAAGG-3’,(下划线示出BsaI酶切位点);
R(Pps-GG3-1):
5’-AGCGTGGGTCTCGTCTTCACTCCATCCACTCCAAGCTC-3’,
(下划线示出BsaI酶切位点)。
以pYLgRNA-AtU3d(GenBank:KR029099)质粒为模板进行第一轮扩增(所用引物:F1(U-F)、R2(gR-R)、R1(AtU3d-R2)、F2(gRTR2)),第二轮扩增用引物F(Pps-GG2-2)和R(Pps-GG3-1)。扩增片段直接连接到ZT4-blunt载体(购自庄盟生物,货号ZC205)载体,并经DNA测序验证。得到的质粒经BsaI酶切分离片段:
3)gRNA表达盒3(目标序列R3):AtU6-1::gRNA-R3
所设计的PCR引物:
F1(U-F):5’-CTCCGTTTTACCTGTGGAATCG-3’;
R2(gR-R):5’-CGGAGGAAAATTCCATCCAC-3’;
R1(AtU6-1-R3):
5’-AGTACCGCAACCGTGAAGAACAATCACTACTTCGTCT-3’;
F2(g RTR3):
5’-TTCTTCACGGTTGCGGTACTGTTTTAGAGCTAGAAAT-3’;
F(Pps-GG3-2):
5’-TTCAGAGGTCTCTAAGACTTTGGAATCGGCAGCAAAGG-3’,(下划线示出BsaI酶切位点);
R(Pgs-GG4):
5’-AGCGTGGGTCTCGAGTCCTTTCCATCCACTCCAAGCTC-3’,(下划线示出BsaI酶切位点)。
以pYLgRNA-AtU6-1(GenBank:KR029101)质粒为模板进行第一轮扩增(所用引物:F1(U-F)、R2(gR-R)、R1(AtU6-1-R3)、F2(gRTR3)),第二轮扩增用引物F(Pps-GG3-2)和R(Pgs-GG4)。扩增片段直接连接到ZT4-blunt载体(购自庄盟生物,货号ZC205)载体,并经DNA测序验证。得到的质粒经BsaI酶切分离片段:
4)gRNA表达盒4(目标序列R4):AtU6-29::gRNA-R4
所设计的PCR引物:
F1(U-F):5’-CTCCGTTTTACCTGTGGAATCG-3’;
R2(gR-R):5’-CGGAGGAAAATTCCATCCAC-3’;
R1(AtU6-29-R4):
5’-TCCGTATAATATTACCGGATCAATCTCTTAGTCGACT-3’;
F2(gRTR4):
5’-ATCCGGTAATATTATACGGAGTTTTAGAGCTAGAAAT-3’;
F(Pps-GG4):
5’-TTCAGAGGTCTCTGACTACATGGAATCGGCAGCAAAGG-3’,(下划线示出BsaI酶切位点);
R(Pgs-GGR):
5’-AGCGTGGGTCTCGACCGATCCATCCACTCC AAGCTC-3’,(下划线示出BsaI酶切位点,粗下划线示出MluI酶切位点,双下划线示出SpeI酶切位点)。
以pYLgRNA-AtU6-29(GenBank:KR029102)质粒为模板进行第一轮扩增(所用引物:F1(U-F)、R2(gR-R)、R1(AtU6-29-R4)、F2(gRTR4)),第二轮扩增用引物F(Pps-GG4)和R(Pgs-GGR)。扩增片段直接连接到ZT4-blunt载体(购自庄盟生物,货号ZC205)载体,并经DNA测序验证。得到的质粒经BsaI酶切分离片段:
3、将上述得到的4种经BsaI酶切分离的片段连接到载体pYLCRISPR/cas9P35S-N(GenBank:KR029112)中,其中4种gRNA表达盒的连接顺序(5’至3’方向)如下:
实施例7:构建具有2个与TYLVC病毒正链DNA互补的gRNA和2个与TYLVC病毒负链DNA互补的gRNA的CRISPR/Cas9表达载体
1、对于CRISPR/Cas9系统中的gRNA,选定TYLVC病毒的目标序列如下:
1)F1:5’-CATTTCCACGCCCGTCTCGAAGG-3’,位于TYLCV基因组的V1基因;
2)F2:5’-GCTGCTGTCCCCATTGTCCAAGG-3’,位于TYLCV基因组的V1基因;
3)R3:5’-TTCTTCACGGTTGCGGTACTGGG-3’,位于TYLCV基因组的V1基因;
4)R4:5’-ATCCGGTAATATTATACGGATGG-3’,位于TYLCV基因组的大间隔区(large intergenic region,LIR)。
2、CRISPR/Cas9载体中的各gRNA表达盒的PCR扩增及克隆
每个gRNA表达盒均采用重叠PCR方法扩增并克隆到T载体,重叠PCR扩增方法如图2所示进行。
1)gRNA表达盒1(目标序列F1):AtU3b::gRNA-F1
所设计的PCR引物:
F1(U-F):5’-CTCCGTTTTACCTGTGGAATCG-3’;
R2(gR-R):5’-CGGAGGAAAATTCCATCCAC-3’;
R1(AtU3b-F1):
5’-TCGAGACGGGCGTGGAAATGTGACCAATGTTGCTCC-3’;
F2(gRTF1):
5’-CATTTCCACGCCCGTCTCGAGTTTTAGAGCTAGAAAT-3’;
F(Pps-GGL-1):
5’-TTCAGAGGTCTCTCTCGATGGAATCGGCAGCAAAGG-3’,(下划线示出BsaI酶切位点,双下划线示出SpeI酶切位点);
R(Pps-GG2-1):
5’-AGCGTGGGTCTCGTCAGGGTCCATCCACTCCAAGCTC-3’,(下划线示出BsaI酶切位点)。
以pYLgRNA-AtU3b(GenBank:KR029097)质粒为模板进行第一轮扩增(所用引物:F1(U-F)、R2(gR-R)、R1(AtU3b-F1)、F2(gRTF1)),第二轮扩增用引物F(Pps-GGL-1)和R(Pps-GG2-1)。扩增片段直接连接到ZT4-blunt载体(购自庄盟生物,货号ZC205),并经DNA测序验证。得到的质粒经BsaI酶切分离片段:
2)gRNA表达盒2(目标序列F2):AtU3d::gRNA-F2
所设计的PCR引物:
F1(U-F):5’-CTCCGTTTTACCTGTGGAATCG-3’;
R2(gR-R):5’-CGGAGGAAAATTCCATCCAC-3’;
R1(AtU3d-F2):
5’-TGGACAATGGGGACAGCAGCTGACCAATGGTGCTTTG-3’;
F2(gRTF2):
5’-GCTGCTGTCCCCATTGTCCAGTTTTAGAGCTAGAAAT-3’;
F(Pps-GG2-2):
5’-TTCAGAGGTCTCTCTGACACTGGAATCGGCAGCAAAGG-3’,(下划线示出BsaI酶切位点);
R(Pps-GG3-1):
5’-AGCGTGGGTCTCGTCTTCACTCCATCCACTCCAAGCTC-3’,(下划线示出BsaI酶切位点)。
以pYLgRNA-AtU3d(GenBank:KR029099)质粒为模板进行第一轮扩增(所用引物:F1(U-F)、R2(gR-R)、R1(AtU3d-F2)、F2(gRTF2)),第二轮扩增用引物F(Pps-GG2-2)和R(Pps-GG3-1)。扩增片段直接连接到ZT4-blunt载体(购自庄盟生物,货号ZC205)载体,并经DNA测序验证。得到的质粒经BsaI酶切分离片段:
3)gRNA表达盒3(目标序列R3):AtU6::gRNA-R3
所设计的PCR引物:
F1(U-F):5’-CTCCGTTTTACCTGTGGAATCG-3’;
R2(gR-R):5’-CGGAGGAAAATTCCATCCAC-3’;
R1(AtU6-1-R3):
5’-AGTACCGCAACCGTGAAGAACAATCACTACTTCGTCT-3’;
F2(gRTR3):
5’-TTCTTCACGGTTGCGGTACTGTTTTAGAGCTAGAAAT-3’;
F(Pps-GG3-2):
5’-TTCAGAGGTCTCTAAGACTTTGGAATCGGCAGCAAAGG-3’,(下划线示出BsaI酶切位点);
R(Pgs-GG4):
5’-AGCGTGGGTCTCGAGTCCTTTCCATCCACTCCAAGCTC-3’,(下划线示出BsaI酶切位点)。
以pYLgRNA-AtU6-1(GenBank:KR029101)质粒为模板进行第一轮扩增(所用引物:F1(U-F)、R2(gR-R)、R1(AtU6-1-R3)、F2(gRTR3)),第二轮扩增用引物F(Pps-GG3-2)和R(Pgs-GG4)。扩增片段直接连接到ZT4-blunt载体(购自庄盟生物,货号ZC205)载体,并经DNA测序验证。得到的质粒经BsaI酶切分离片段:
4)gRNA表达盒4(目标序列R4):AtU6-29::gRNA-R4
所设计的PCR引物:
F1(U-F):5’-CTCCGTTTTACCTGTGGAATCG-3’;
R2(gR-R):5’-CGGAGGAAAATTCCATCCAC-3’;
R1(AtU6-29-R4):
5’-TCCGTATAATATTACCGGATCAATCTCTTAGTCGACT-3’;
F2(gRTR4):
5’-ATCCGGTAATATTATACGGAGTTTTAGAGCTAGAAAT-3’;
F(Pps-GG4):
5’-TTCAGAGGTCTCTGACTACATGGAATCGGCAGCAAAGG-3’,(下划线示出BsaI酶切位点);
R(Pgs-GGR):
5’-AGCGTGGGTCTCGACCGATCCATCCACTCCAAGCTC-3’,(下划线示出BsaI酶切位点,粗下划线示出MluI酶切位点,双下划线示出SpeI酶切位点)。
以pYLgRNA-AtU6-29(GenBank:KR029102)质粒为模板进行第一轮扩增(所用引物:F1(U-F)、R2(gR-R)、R1(AtU6-29-R4)、F2(gRTR4)),第二轮扩增用引物F(Pps-GG4)和R(Pgs-GGR)。扩增片段直接连接到ZT4-blunt载体(购自庄盟生物,货号ZC205)载体,并经DNA测序验证。得到的质粒经BsaI酶切分离片段:
3、将上述得到的4种经BsaI酶切分离的片段连接到载体pYLCRISPR/cas9P35S-N(GenBank:KR029112)中,其中4种gRNA表达盒的连接顺序(5’至3’方向)如下:
实施例8:构建具有2个与TYLVC病毒负链DNA互补的gRNA和2个与TYLVC病毒正链DNA互补的gRNA的CRISPR/Cas9表达载体
1、对于CRISPR/Cas9系统中的gRNA,选定TYLVC病毒的目标序列如下:
1)R1:5’-CTTCGGCGAACCTTCGAGACGGG-3’,位于TYLCV基因组的V1基因;
2)R2:5’-GGACAATGGGGACAGCAGCACGG-3’,位于TYLCV基因组的V1基因;
3)F3:5’-TCTTCTTGGTTCGTGATAGAAGG-3’,位于TYLCV基因组的V1基因;
4)F4:5’-ATCCGTATAATATTACCGGATGG-3’,位于TYLCV基因组的大间隔区(large intergenic region,LIR)。
2、CRISPR/Cas9载体中的各gRNA表达盒的PCR扩增及克隆
每个gRNA表达盒均采用重叠PCR方法扩增并克隆到T载体,重叠PCR扩增方法如图2所示进行。
1)gRNA表达盒1(目标序列R1):AtU3b::gRNA-R1
所设计的PCR引物:
F1(U-F):5’-CTCCGTTTTACCTGTGGAATCG-3’;
R2(gR-R):5’-CGGAGGAAAATTCCATCCAC-3’;
R1(AtU3b-R1):
5’-GTCTCGAAGGTTCGCCGAAGTGACCAATGTTGCTCC-3’;
F2(gRTR1):
5’-CTTCGGCGAACCTTCGAGACGTTTTAGAGCTAGAAAT-3’;
F(Pps-GGL-1):
5’-TTCAGAGGTCTCTCTCGATGGAATCGGCAGCAAAGG-3’,(下划线示出BsaI酶切位点,双下划线示出SpeI酶切位点);
R(Pps-GG2-1):
5’-AGCGTGGGTCTCGTCAGGGTCCATCCACTCCAAGCTC-3’,(下划线示出BsaI酶切位点)。
以pYLgRNA-AtU3b(GenBank:KR029097)质粒为模板进行第一轮扩增(所用引物:F1(U-F)、R2(gR-R)、R1(AtU3b-R1)、F2(gRTR1)),第二轮扩增用引物F(Pps-GGL-1)和R(Pps-GG2-1)。扩增片段直接连接到ZT4-blunt载体(购自庄盟生物,货号ZC205)载体,并经DNA测序验证。得到的质粒经BsaI酶切分离片段:
2)gRNA表达盒2(目标序列R2):AtU3d::gRNA-R2
所设计的PCR引物:
F1(U-F):5’-CTCCGTTTTACCTGTGGAATCG-3’;
R2(gR-R):5’-CGGAGGAAAATTCCATCCAC-3’;
R1(AtU3d-R2):
5’-TGCTGCTGTCCCCATTGTCCTGACCAATGGTGCTTTG-3’;
F2(gRTR2):
5’-GGACAATGGGGACAGCAGCAGTTTTAGAGCTAGAAAT-3’;
F(Pps-GG2-2):
5’-TTCAGAGGTCTCTCTGACACTGGAATCGGCAGCAAAGG-3’,(下划线示出BsaI酶切位点);
R(Pps-GG3-1):
5’-AGCGTGGGTCTCGTCTTCACTCCATCCACTCCAAGCTC-3’,(下划线示出BsaI酶切位点)。
以pYLgRNA-AtU3d质粒(GenBank:KR029099)为模板进行第一轮扩增(所用引物:F1(U-F)、R2(gR-R)、R1(AtU3d-R2)、F2(gRTR2)),第二轮扩增用引物F(Pps-GG2-2)和R(Pps-GG3-1)。扩增片段直接连接到ZT4-blunt载体(购自庄盟生物,货号ZC205)载体,并经DNA测序验证。得到的质粒经BsaI酶切分离片段:
3)gRNA表达盒3(目标序列F3):AtU6-1::gRNA-F3
所设计的PCR引物:
F1(U-F):5’-CTCCGTTTTACCTGTGGAATCG-3’;
R2(gR-R):5’-CGGAGGAAAATTCCATCCAC-3’;
R1(AtU6-1-F3):5’
TCTATCACGAACCAAGAAGACAATCACTACTTCGTCT-3’;
F2(gRTF3):
5’-TCTTCTTGGTTCGTGATAGAGTTTTAGAGCTAGAAAT-3’;
F(Pps-GG3-2):
5’-TTCAGAGGTCTCTAAGACTTTGGAATCGGCAGCAAAGG-3’,(下划线示出BsaI酶切位点);
R(Pgs-GG4):
5’-AGCGTGGGTCTCGAGTCCTTTCCATCCACTCCAAGCTC-3’,(下划线示出BsaI酶切位点)。
以pYLgRNA-AtU6-1(GenBank:KR029101)质粒为模板进行第一轮扩增(所用引物:F1(U-F)、R2(gR-R)、R1(AtU6-1-F3)、F2(gRTF3)),第二轮扩增用引物F(Pps-GG3-2)和R(Pgs-GG4)。扩增片段直接连接到ZT4-blunt载体(购自庄盟生物,货号ZC205)载体,并经DNA测序验证。得到的质粒经BsaI酶切分离片段:
4)gRNA表达盒4(目标序列F4):AtU6-29::gRNA-F4
F1(U-F):5’-CTCCGTTTTACCTGTGGAATCG-3’;
R2(gR-R):5’-CGGAGGAAAATTCCATCCAC-3’;
R1(AtU6-29-F4):
5’-TCCGGTAATATTATACGGATCAATCTCTTAGTCGACT-3’;
F2(gRTF4):
5’-ATCCGTATAATATTACCGGAGTTTTAGAGCTAGAAAT-3’;
F(Pps-GG4):
5’-TTCAGAGGTCTCTGACTACATGGAATCGGCAGCAAAGG-3’,(下划线示出BsaI酶切位点);
R(Pgs-GGR):
5’-AGCGTGGGTCTCGACCGATCCATCCACTCCAAGCTC-3’,(下划线示出BsaI酶切位点,粗下划线示出MluI酶切位点,双下划线示出SpeI酶切位点)。
以pYLgRNA-AtU6-29(GenBank:KR029102)质粒为模板进行第一轮扩增(所用引物:F1(U-F)、R2(gR-R)、R1(AtU6-29-F4)、F2(gRTF4)),第二轮扩增用引物F(Pps-GG4)和R(Pgs-GGR)。扩增片段直接连接到ZT4-blunt载体(购自庄盟生物,货号ZC205)载体,并经DNA测序验证。得到的质粒经BsaI酶切分离片段:
3、将上述得到的4种经BsaI酶切分离的片段连接到载体pYLCRISPR/cas9P35S-N(GenBank:KR029112),其中4种gRNA表达盒的连接顺序(5’至3’方向)如下:
实施例9:将核酸内切酶系统的表达载体转入农杆菌
1)农杆菌感受态细胞的制备
取-70℃保存的农杆菌LBA4404(参见文献“Hoekema,A.,Hirsch,P.R.,Hooykaas,P.J.J.,&Schilperoort,R.A.1983.A binary plant vector strategy based on separation of vir-and T-region of the Agrobacterium tumefaciens Ti-plasmid.Nature,303:179-180”)划线于含有50mg/L利福平的LB(每升含胰化蛋白胨10g,酵母提取物5g,NaCl 10g,琼脂12g,pH7.0)平板上,28℃培养2-3天。挑取单菌落,28℃培养于50ml含50mg/L利福平的液体LB(每升含胰化蛋白胨10g,酵母提取物5g,NaCl 10g,pH7.0)中,150rpm振荡培养过夜,培养至OD600=0.5。将农杆菌菌液转入无菌50ml离心管,4℃,4000rpm离心4分钟,弃上清液,加入5ml预冷0.15mol/L NaCl悬浮细胞,4℃,4000rpm离心4分钟,去上清液;再用0.15mol/L NaCl悬浮细胞,4000rpm离心4分钟,去上清液;加入2ml预冷20mmol/L CaCl2溶液,轻轻悬浮细胞,冰上放置30分钟,加入终浓度为15%(v/v)的甘油,混匀后每管分装100μl,液氮冷冻后存于-70℃保存。
2)载体质粒转化农杆菌
取-70℃保存农杆菌感受态细胞,置于冰水中解冻,然后取1μg左右的实施例1构建的载体质粒DNA加入到100μl所制备的农杆菌感受态细胞中,混匀后,冰浴30分钟;液氮冷冻1分钟,取出后37℃水浴2分钟,冰浴2分钟;再次液氮冷冻1分钟,取出后37℃水浴2分钟,冰浴2分钟;加入1ml液体LB(pH7.0),于28℃、150rpm震荡培养约3小时。离心收集菌体,100μl液体LB重悬农杆菌,并涂在含50mg/L卡那霉素、50mg/L利福平的LB(pH7.0)平板上,在28℃经2-3天的培养直到形成单菌落。取单菌落到含有50mg/L卡那霉素、50mg/L利福平的液体LB(pH7.0)中,于28℃,150rpm振荡培养16小时。取2μl菌液进行PCR验证,阳性克隆农杆菌液保存备用。
实施例10:用农杆菌转化番茄无菌苗子叶或下胚轴外植体,获得转基因番茄
1)番茄的农杆菌转化
番茄种子用70%乙醇浸泡30秒,然后用10%(v/v)次氯酸钠消毒15分钟,期间不断摇动,随后用无菌水冲洗3-5遍,接种于1/2MS培养基(MS培养基的大量元素用量减半)上培养10天。取番茄无菌苗的子叶或下胚轴为外植体,用OD600=0.3左右的实施例9制备的农杆菌菌液浸泡感染外植体5分钟。取出外植体,置于无菌干燥滤纸上吸干残留菌液,转入MS+0.5mg/L IAA(吲哚乙酸)+2.0mg/L 6-BA(6-苄氨基腺嘌呤)培养基(pH5.8)上黑暗处共培养48小时,然后转入MS+0.5mg/L IAA+2.0mg/L 6-BA+150mg/L羧苄青霉素+100mg/L头孢霉素+50mg/L卡那霉素筛选培养基(pH5.8)上,于25℃、光照18小时/黑暗6小时的光照周期进行培养,每三周换一次培养基。约40天左右分化出绿色芽点;待再生芽长到3-4cm时切下并转入1/2MS(MS培养基的大量元素用量减半)+1.0mg/L IBA+100mg/L头孢霉素+150mg/L羧苄青霉素+50mg/L卡那霉素生根培养基(pH5.8)中培养;根系发达后移栽至土壤。
2)转基因番茄外源DNA插入的鉴定
番茄植株总DNA提取(CTAB法,即十六烷基三甲基溴化铵法):取0.1-0.2g新鲜叶片,液氮研磨成粉末,转入1.5ml离心管,加入600μl预热CTAB裂解液(2g CTAB,8.18g NaCl,0.74g EDTA.Na2.2H2O,加入10ml 1mol/L的Tris-HCL(pH8.0),0.2ml巯基乙醇,加水定容到100ml),涡旋混匀,65℃水浴1小时,加入600μl氯仿:异戊醇(体积比24:1),颠倒50次混匀,10000rpm离心15分钟,取上清于新1.5ml离心管,加入等体积的预冷异丙醇,充分混匀,-20℃放置30分钟。12000rpm离心15分钟,弃上清,加入1ml 75%(v/v)酒精洗涤DNA沉淀,弃酒精,风干沉淀。将沉淀溶于加入含RNase A(购自生工生物工程(上海)股份有限公司,货号:B100675)10mg/mL的100μl TE缓冲液(pH 8.0)中,37℃保温30分钟,加入100μl的酚/氯仿(体积比1:1),混匀,12000rpm离心10分钟,取上清。加入1/10体积的3mol/L NaAc和2.5倍体积的无水乙醇,室温放置10分钟,12000g离心10分钟,弃上清。用70%(v/v)乙醇冲洗后风干沉淀,溶于500μl灭菌ddH2O中。DNA样品保存在-20℃下备用。
外源插入DNA的PCR分析:用转入番茄的外源基因的特异引物进行扩增,外源基因包括卡那霉素抗性基因(引物:NPTII-F:5’-TCTCATGCTGGAGTTCTTCGC-3’;NPTII-R:5’-GTCACCGACTTGAGCCATTTG-3’)和目的基因(引物以具体实施事例而定,引物长度约20个碱基左右,Tm值55℃)。
在本实施例中,利用上述卡那霉素抗性基因引物,以转基因再生番茄植株DNA为模板进行扩增,扩增产物大小为0.5kb。PCR扩增程序如下:95℃预变性5分钟;94℃变性30秒,56℃退火30秒,72℃延伸50秒,30个循环;最后再经72℃延伸5分钟。PCR产物经1%琼脂糖凝胶电泳,电泳检测结果显示出预期0.5kb大小的片段。该片段经DNA测序验证。
3)转基因番茄外源基因表达的鉴定
番茄植株总RNA提取(Trizol试剂法):取0.1-0.2g新鲜叶片,液氮研磨成粉末,转入1.5ml离心管,立即加入1ml Trizol试剂(购自Invitrogen,货号:15596-018),盖紧盖子,充分剧烈振荡15秒并于室温静置5分钟,于2-8℃12000g离心15分钟。离心后样品分层,上层水相中含RNA,下层有机相中含蛋白和DNA。吸取上层转入1.5ml离心管,加入等体积异丙醇,轻轻混匀,于室温静置10分钟后,于2-8℃12000g离心10分钟,弃去上清,加入1ml 75%(v/v)乙醇,轻轻混匀,2-8℃7500g离心5分钟,弃上清。沉淀RNA样品凉干,加入50μl用DEPC(diethypyrocarbonate,焦碳酸二乙酯)处理的纯净水溶解。RNA样品保存在-70℃下备用。
外源基因CRISPR/Cas9表达的RT-PCR分析:用商业性试剂盒(购自TAKARA公司,货号:RR064A),按照制造商的说明,将RNA反转录得到cDNA。用cDNA作为模板,用CRISPR/Cas9基因的特异引物(RT-Cas9-F:5’-ACCCAGTTGAGAACACCC-3’;RT-Cas9-R:5’-CTTAGCGGTAGCCTTTCC-3’)进行PCR扩增,扩增产物大小为0.7kb。PCR扩增程序如下:95℃预变性5分钟;94℃变性30秒,56℃退火30秒,72℃延伸50秒,35个循环;最后再经72℃延伸5分钟。PCR产物经1%琼脂糖凝胶电泳,电泳检测结果显示出预期0.7kb大小的片段。该片段经DNA测序验证。
实施例11:番茄抗TYLCV病毒的农杆菌侵染接种鉴定
1)构建TYLCV侵染农杆菌用克隆:设计如下引物:
TY-F:5’-GTCTGTCTTGCAATATGTGGGATCC-3’
TY-R:5’-GGAAATTCATTTAGAAGTGGATCCCAC-3’
用本实验室分离克隆的TYLCV基因组(GenBank:KC211184.1,SEQ ID No.1)作为模板,扩增出病毒DNA片段,克隆到pMD18-T载体,然后,用BamHI和SacI进行酶切,分离出0.6kb的病毒DNA片段,克隆到pGreenII 0029(http://www.pgreen.ac.uk/)载体的BamHI和SacI位点。用BamHI在上述pMD18-T载体克隆中切下全长病毒DNA,克隆到上述含有0.6Kb DNA的pGreenII 0029质粒的BamHI位点中,得到病毒侵染克隆。
2)将侵染克隆转入农杆菌:用实施例9所述的农杆菌转化方法将侵染克隆转入农杆菌C58C1(参见文献“Van Larebeke,N.,G.Engler,M.Holsters,S.Van den Elsacker,I.Zaenen,R.A.Schilperoort,and J.Schell.1974.Large plasmid in Agrobacterium tumefaciens essential for crown gall inducing ability.Nature,252:169-170”),得到用于侵染番茄的农杆菌。
3)侵染用农杆菌的准备:将上述农杆菌接种到50ml LB液体培养基(每升含胰化蛋白胨10g,酵母提取物5g,NaCl 10g,50mg/L利福平和50mg/L卡那霉素,pH7.0)中,28℃下,150rpm振荡培养过夜。菌液浓度为OD600=0.6左右时离心收集菌体,然后将菌体悬浮在侵染液(10mmol/L MgCl2,10mM 2-吗啉乙磺酸(2-morpholinoethanesulfonic acid,购自Sigma-Aldrich,货号:M3671),pH 5.2)中,使菌液浓度为OD600=2.0左右。使用前加入乙酰丁香酮(购自Sigma-Aldrich,货号:D134406)至200μmol/L。
4)番茄枝条的侵染:切取约6cm长的番茄幼枝,放入含上述农杆菌液的烧杯中,将烧杯放入置于真空容器内,抽真空(-0.09MPa)5分钟。真空完毕后,取出侵染枝条,插入营养土中生根长芽(25℃),30天观察长根植株发病情况。图5示出野生型经侵染后出现病毒感染症状。图6示出如实施例10所述制备的转基因植株经侵染后明显未出现病毒病症状。
实施例12:携带TYLCV的烟粉虱接种鉴定
将如实施例10所述制备的转基因番茄植株及其非转基因对照置于防虫温室,25℃、光照16小时/黑暗8小时的光照周期进行培养,通过携带TYLCV的烟粉虱进行捕食和传毒,30天后观察植株症状。图7示出野生型番茄经烟粉虱接种后出现病毒感染症状。图8示出转基因植株经烟粉虱接种后明显未出现病毒病症状。
将植株种植于大田,结果发现非转基因植株严重感病,植株上半部分的新叶明显卷曲皱缩,植株矮化,生长期后期开花不畅(图9);而转基因植株不受病害,生长结实正常(图10)。
实施例13:番茄接种TYLCV后,植株体内病毒的Southern杂交分析
在实施例12和13中,番茄是否感病,从植株的表型可以判断,如植株顶端新叶出现扭曲、变黄节间缩短等,表明植株感病,反之,接种病毒后,生长如同未接种病毒植株,表明是培育得到的抗病毒番茄。然而,为更好地说明问题,可以采用分子检测手段,分析植株体内是否含有TYLCV病毒DNA,用Southern杂交分析(参见文献“Southern,E.M.1975.Detection of specific sequences among DNA fragments separated by gel electrophoresis.Journal of molecular biology,98:503-517”)是通常采用的有效技术手段。
用实施例10所述番茄植株DNA的提取方法(CTAB法)提取接种TYLCV病毒的番茄叶片总DNA,DNA经琼脂糖电泳分开,用10倍SSC(saline sodium citrate)缓冲溶液转移至硝酸纤维素膜上。用地高辛标记的C1基因DNA探针,利用杂交试剂盒(购自罗氏(Roche)公司,货号:11175041910),按照制造商推荐的操作方法进行杂交,用百乐(BIO-RAD)的ChemiDoc XRS检测并拍照。
实施例14
重复实施例9-13所述的方法,不同之处在于用实施例2制备的载体质粒DNA转化农杆菌,然后转化番茄,同样获得了抗TYLCV病毒的番茄植株。
实施例15-20
按照实施例14所述方法进行实验操作,不同之处在于分别用实施例3-8制备的载体质粒DNA转化农杆菌,然后转化番茄,同样获得了抗TYLCV病毒的番茄植株。
- 还没有人留言评论。精彩留言会获得点赞!