氮取代3‑氧代‑6‑取代四氢喹喔啉结构化合物、其制备方法及其医药用途与流程
本发明属于医药领域,具体涉及一种氮取代3-氧代-6-取代四氢喹喔啉结构化合物、其制备方法及其医药用途。
背景技术:
微管是由α,β微管蛋白异二聚体聚合而成的管状聚合物,在细胞中具有诸多功能,特别是以纺锤体的形式参与细胞的有丝分裂过程。肿瘤细胞具有快速增殖能力,若抑制其纺锤体的形成,必将导致有丝分裂的阻断,使得肿瘤细胞生长停滞于G2/M期,抑制细胞增殖。
微管以胞质微管和纺锤体微管两种形式存在,参与细胞运动、细胞器定位、胞内物质运输及真核细胞的有丝分裂过程等。细胞进入分裂期,其胞质微管解聚成微管蛋白后再组装成纺锤体微管。在分裂后期,纺锤体微管牵引着姐妹染色单体从赤道面移向纺锤体两极。在有丝分裂结束后,纺锤体微管解聚再组装成胞质微管。因此,这种微管蛋白/微管之间的动态转换(聚合和解聚),为细胞正常有丝分裂所必需。若破坏此循环将致使进入有丝分裂的细胞停止分裂,进而延长细胞周期或诱导细胞凋亡。
康普瑞汀(Combretastatin A4)是从风车子属南非柳树Combretum caffrum中分离得到的天然产物,其能够抑制微管蛋白的聚合,具有较好的抗肿瘤活性。然而,其水溶性差及容易发生双键的异构化而生成无活性的反式同分异构体限制了其临床应用。
康普瑞汀的结构改造主要包括:(1)引入磷酸、氨基酸等结构以改善其水溶性;(2)以磺酰胺、羰基、五元芳香杂环等替代双键以解决其双键在体内的不稳定问题。然而,上述所有报道的康普瑞汀衍生物均不同于本发明所涉及的化合物。
技术实现要素:
本发明的目的在于提供一种氮取代3-氧代-6-取代四氢喹喔啉类新颖结构化合物;本发明的另一个目的是提供该类化合物的制备方法;本发明的第三个目的是提供该类化合物或含有该类化合物的药物组合物在制备预防或治疗抗肿瘤或除肿瘤以外的增殖性疾病药物中的应用。
本发明的具体技术方案如下:
作为本发明的第一方面,氮取代3-氧代-6-取代四氢喹喔啉结构化合物以及其任意组合或其药用盐,结构通式如式(Ⅰ):
其中R1代表羟基,链状C1-6的烷氧基,3-6个环成员的环烷氧基,链状C1-6的烷胺基,3-6个环成员的环烷胺基,苯环或取代的苯环基,芳香环或取代的芳香环基,芳香杂环或取代的芳香杂环基,3-6个环成员的含氮和/或含氧杂环基;
R2代表氢,卤素(氟,氯,溴,碘),羟基,链状C1-6的烷氧基;
R3代表氢,卤素(氟,氯,溴,碘),羟基,链状C1-6的烷氧基;
R4代表氢,卤素(氟,氯,溴,碘),羟基,链状C1-6的烷氧基;
W代表亚甲基,羰基,磺酰基,亚磺酰基。
优选地,R1为羟基、甲氧基、正丁胺基、环己胺基、吗啉基或苯胺基;R2为氢或甲氧基;R3为氢、氟或甲氧基;R4为氢或甲氧基;W为亚甲基、羰基或亚甲基羰基。
本发明的化合物可按照常规方法制备为药用盐形式;包括其有机酸盐及无机酸盐:有机酸包括(但不限于)乙酸、马来酸、富马酸、酒石酸、琥珀酸、乳酸、对甲苯磺酸、水杨酸、草酸等,无机酸包括(但不限于)盐酸、硫酸、磷酸、二磷酸、氢溴酸、硝酸等。含有羧酸化合物的金属盐:包括(但不限于)钾盐、钠盐等。
作为本发明的第二个方面,本发明的化合物可以用以下方法制备得到,其合成路线如下:
具体步骤为:
(1)制备3-硝基-4-氨基苯甲酸酯或3-硝基-4-氨基苯甲酰胺(2a-2e)
以3-硝基-4-氨基苯甲酸(即化合物1)为起始原料,在二氯亚砜作用下与甲醇脱水缩合,得到化合物2a;以3-硝基-4-氨基苯甲酸(即化合物1)为起始原料,二氯甲烷为溶剂,在1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐(EDCI)作用下与不同的胺类化合物缩合得到化合物2b-2e;
(2)制备3-硝基-4-((2-乙氧基-2-氧代乙基)氨基)苯甲酸酯或3-硝基-4-((2-乙氧基-2-氧代乙基)氨基)苯甲酰胺(3a-4e)
化合物2a-2e分别以碳酸铯为碱,与溴乙酸乙酯发生亲核取代反应,得到化合物3a-3e;
(3)制备3-氧代-1,2,3,4-四氢喹喔啉-6-羧酸酯或3-氧代-1,2,3,4-四氢喹喔啉-6-羧酰胺(4a-4e)
化合物3a-3e分别以甲醇为溶剂,以钯碳为催化剂,在氢气作用下经还原、环合一锅法得到化合物4a-4e;
(4)制备1-取代苄基-3-氧代-1,2,3,4-四氢喹喔啉-6-羧酸酯(5a-5c)
化合物4a以乙腈为溶剂,以碳酸钾为碱、碘化钾为催化剂与不同取代的氯化苄发生亲核取代反应得到化合物5a-5c;
(5)制备1-(3,4,5-三甲氧苄基)-3氧代-1,2,3,4-四氢喹喔啉-6-羧酸(6)
化合物5a以四氢呋喃/水为溶剂,在氢氧化锂作用下水解,之后用浓盐酸酸化处理得到化合物6;
(6)制备1-(3,4,5-三甲氧苄基)-3氧代-1,2,3,4-四氢喹喔啉-6-羧酰胺(7a-7b)
化合物6以甲苯为溶剂,与草酰氯作用生成中间体,该中间体以四氢呋喃为溶剂,碳酸钾为碱,分别与不同的苯胺作用生成化合物7a-7b;
(7)制备取代苯甲酰氯或取代苯乙酰氯(9a-9e)
原料8a-8e分别与二氯亚砜反应生成化合物9a-9e;
(8)制备1-取代苯甲酰基-3-氧代-1,2,3,4-四氢喹喔啉-6-羧酸酯、1-取代苯甲酰基-3-氧代-1,2,3,4-四氢喹喔啉-6-羧酰胺或1-取代苯乙酰基-3-氧代-1,2,3,4-四氢喹喔啉-6-羧酸酯(10a-10l)
化合物4a-4e以四氢呋喃为溶剂、碳酸钾为碱分别与化合物9a-9e反应得到化合物10a-10l。
作为本发明的第三个方面,还提供了上述任一化合物以及其任意组合或其药用盐,在制备预防或治疗抗肿瘤疾病或其它增殖性疾病药物中的应用。
本发明还提供了一种药物组合物,通式(Ⅰ)化合物、药学上可接受的盐或溶剂化物可以添加药学上可接受的载体制成常见的药用制剂,如片剂、胶囊、粉剂、糖浆、液剂、悬浮剂、针剂,可以加入香料、甜味剂、液体或固体填充料或稀释剂等常用的药物辅料。
为评价本发明合成化合物及其药物组合物的抗肿瘤活性,对本发明的化合物进行了细胞和分子水平的药理活性测试。
本发明化合物及其药物组合物经体外细胞生长抑制实验,结果显示该类化合物普遍具有较强的细胞生长抑制作用。化合物5c、10c对细胞生长抑制作用的半数抑制浓度(IC50)达到几微摩尔/升(μM),化合物10d达到纳摩尔水平(nM)。这些化合物的细胞生长抑制活性明显优于阿霉素。
本发明化合物及其药物组合物经体外微管蛋白聚合抑制实验,结果显示化合物5c、10c及10d在20μM具有明显的抑制微管蛋白聚合抑制作用,化合物10d对微管蛋白聚合抑制作用的半数抑制浓度(IC50)为3.9μM。
本发明化合物及其药物组合物经体外细胞生长周期实验,结果显示化合物10d在0.15μM能够明显地将细胞阻滞在G2/M期。
本发明所述化合物和药学上可接受的载体在临床上可以采用口服、静脉注射等给药方式。在临床上可以用于单药治疗,也可以和其他临床上使用的化疗药物及放疗等治疗手段联合用于前述癌症的治疗。
附图说明
图1是体外微管蛋白聚合抑制作用图。其中A为空白对照、紫杉醇(3μM)、康普瑞汀(2.5μM)、化合物5c(20μM)、10c(20μM)及10d(20μM)对微管蛋白聚合影响;B为化合物10d在1μM,5μM,10μM,50μM浓度时对微管蛋白聚合抑制作用。
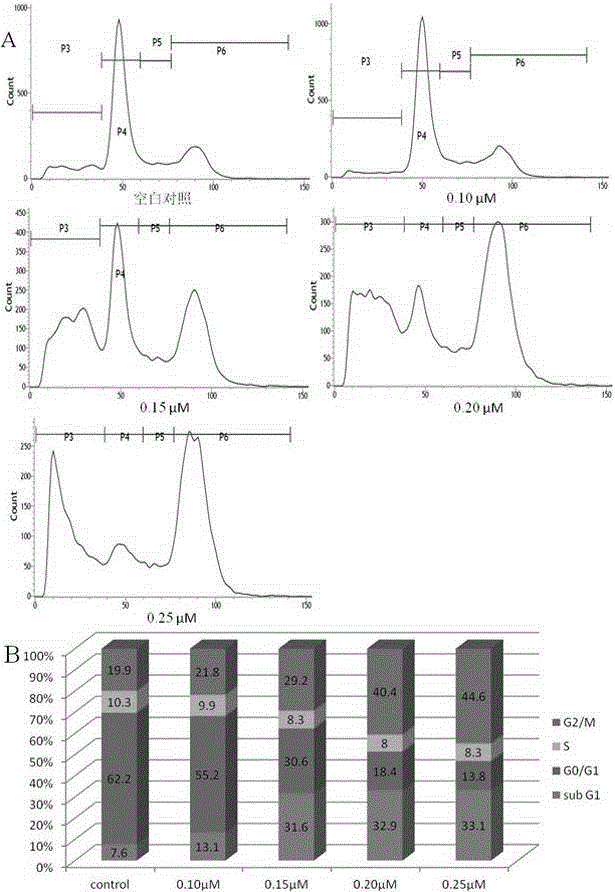
图2是化合物10d对HeLa细胞生长周期影响图。其中A为空白对照、化合物10d在0.1μM,0.15μM,0.20μM,0.25μM浓度时对细胞生长周期影响;B为处于不同生长期细胞统计图。
具体实施方式
以下结合具体实施例对本发明的技术方案做进一步详细说明,但本发明的保护范围并不局限于此。下述实施例中柱层析所用洗脱剂的比例均为体积比。
实施例1
1-(4-甲氧苄基)-3-氧代-1,2,3,4-四氢喹喔啉-6-甲酸甲酯(5a)的制备
(1)3-硝基-4-氨基苯甲酸甲酯的制备(2a)
将3-硝基-4-氨基苯甲酸(即化合物1,4.50g,24.7mmol)、无水甲醇(80mL),依次投入到250mL三颈瓶中,待温度降至0℃,用恒压滴液漏斗慢慢滴加氯化亚砜(5.38mL,74.1mmol,3equiv),滴加完毕,0℃继续搅拌30分钟,升温至50℃反应4h,有大量的黄色固体产生,抽滤,将滤液中的甲醇蒸干,加入30mL水与30mL乙酸乙酯,分液,用无水硫酸钠干燥有机层,蒸干溶剂,得到黄色固体,与前面滤饼合并后干燥,得到化合物2a,黄色固体4.32g,收率89.2%。熔点:181.9-182.7℃。1H NMR(300MHz,CDCl3)δ8.85(d,J=1.9Hz,1H),7.99(dd,J=8.8,1.9Hz,1H),6.83(d,J=8.8Hz,1H),6.42(brs,2H),3.90(s,3H).
(2)3-硝基-4-((2-乙氧基-2-氧代乙基)氨基)苯甲酸甲酯的制备(3a)
将2a(300mg,1.53mmol),碳酸铯(600mg,1.84mmol,1.2equiv),溴乙酸乙酯(1.70mL,15.3mmol,10equiv)依次投入到25mL圆底烧瓶中,升温至140℃反应20h,反应完毕,减压蒸出溴乙酸乙酯,残留物经柱层析(石油醚:乙酸乙酯=20︰1),得252mg黄色固体3a,收率58.3%。熔点:127.6-128.8℃。1H NMR(300MHz,CDCl3)δ8.91(d,J=2.0Hz,1H),8.75(brs,1H),8.09(dd,J=8.9,1.8Hz,1H),6.72(d,J=9.0Hz,1H),4.31(q,J=7.1Hz,2H),4.14(d,J=5.2Hz,2H),3.91(s,3H),1.33(t,J=7.1Hz,3H).
(3)3-氧代-1,2,3,4-四氢喹喔啉-6-羧酸甲酯的制备(4a)
将3a(300mg,1.06mmol),无水甲醇(100mL),10%钯碳(56mg,0.053mmol,0.05equiv,CAS号:64741-65-7,下同)依次投入到250mL圆底烧瓶中,氢气球置换空气三次,室温搅拌反应10小时,反应完毕,抽滤,蒸出溶剂,柱层析(200-300目硅胶,二氯甲烷︰甲醇=80︰1),得到180mg灰白色固体4a,收率82.4%。熔点:大于280℃。1H NMR(300MHz,CDCl3)δ7.74(brs,1H),7.60(d,J=8.1Hz,1H),7.38(s,1H),6.64(d,J=8.2Hz,1H),4.09(s,2H),3.87(s,3H).MS(ESI)[M+H]+207.00.
(4)1-(4-甲氧苄基)-3-氧代-1,2,3,4-四氢喹喔啉-6-甲酸甲酯的制备(5a)
将4a(30mg,0.145mmol),乙腈(2mL),无水碳酸钾(24mg,0.174mmol,1.2equiv),碘化钾(12mg,0.073mmol,0.5equiv),4-甲氧基氯化苄(34mg,0.218mmol,1.5equiv)依次投入到10mL茄型瓶中,升温至70℃反应6小时,反应完毕,蒸出溶剂,柱层析(200-300目硅胶,二氯甲烷︰甲醇=80︰1),得到20mg灰白色固体,收率42.3%。熔点:217-219℃。1H NMR(300MHz,CDCl3)δ8.48(brs,1H),7.65(d,J=8.4Hz,1H),7.44(s,1H),7.19(d,J=8.0Hz,2H),6.88(d,J=8.3Hz,2H),6.76(d,J=8.2Hz,1H),4.44(s,2H),3.92(s,2H),3.87(s,3H),3.80(s,3H).13C NMR(75MHz,CDCl3)δ166.7,165.5,159.3,138.8,128.8,127.1,126.7,125.0,119.9,116.6,114.4,110.9,55.4,52.8,51.9,51.6.HRMS(ESI):calcd for C18H19N2O4[M+H]+327.13393,found 327.13388.
实施例2
1-(3,4-二甲氧苄基)-3-氧代-1,2,3,4-四氢喹喔啉-6-甲酸甲酯(5b)的制备
除第(4)步中用3,4-二甲氧基氯化苄替代4-甲氧基氯化苄外,其他步骤及提纯方法同实施例1,得到29mg白色固体,收率56.1%。熔点:155.9-157.5℃。1H NMR(300MHz,CDCl3)δ9.05(s,1H),7.65(dd,J=8.5,1.8Hz,1H),7.50(d,J=1.8Hz,1H),6.82(s,2H),6.79-6.74(m,2H),4.43(s,2H),3.92(s,2H),3.87(s,6H),3.83(s,3H).13C NMR(75MHz,CDCl3)δ166.8,165.8,149.5,148.7,138.9,127.6,126.6,125.2,120.0,119.9,116.8,111.4,110.9,110.5,56.0,53.2,52.0,51.6.HRMS(ESI):calcd for C19H21N2O5[M+H]+357.14450,found 357.14450.
实施例3
1-(2,3,4-三甲氧苄基)-3-氧代-1,2,3,4-四氢喹喔啉-6-甲酸甲酯(5c)的制备
除第(4)步中用3,4,5-三甲氧基氯化苄替代4-甲氧基氯化苄外,其他步骤及提纯方法同实施例1,得到38mg白色固体,收率67.8%。熔点:188.9-191.0℃。1H NMR(300MHz,CDCl3)δ8.02(brs,1H),7.67(dd,J=8.5,1.9Hz,1H),7.43(d,J=1.9Hz,1H),6.78(d,J=8.6Hz,1H),6.47(s,2H),4.42(s,2H),3.93(s,2H),3.88(s,3H),3.84(s,3H),3.81(s,6H).13C NMR(75MHz,CDCl3)δ166.7,165.7,153.8,138.8,137.5,131.0,126.7,125.1,120.2,116.8,111.0,104.3,60.9,56.2,53.8,52.0,51.8.HRMS(ESI):calcd for C20H23N2O6[M+H]+387.15506,found 387.15512.
实施例4
1-(2,3,4-三甲氧苄基)-3-氧代-1,2,3,4-四氢喹喔啉-6-甲酸(6)的制备
将5c(30mg,0.0776mmol)、四氢呋喃(2mL)、蒸馏水(1mL)和氢氧化锂(3.7mg,0.155mmol,2equiv)依次投入到10mL茄型瓶中,升温至40℃反应10小时,反应完毕,蒸出四氢呋喃,滴加0.5mL浓盐酸(12mol/L),析出白色固体,静置30分钟,抽滤得到21mg灰白色固体,收率72.6%。熔点:135.3-136.3℃。1H NMR(300MHz,CDCl3)δ11.95(brs,1H),7.73(s,1H),7.51(d,J=8.1,1H),6.67(d,J=8.4,1H),6.51(s,2H),4.46(s,2H),4.04(s,2H),3.84(s,9H).13C NMR(75MHz,CDCl3)δ170.3,167.8,153.8,138.9,137.5,131.0,127.2,124.4,119.0,118.6,109.9,104.1,60.9,56.3,53.9,51.3.HRMS(ESI):calcd for C19H21N2O6[M+H]+373.13941,found 373.13940.
实施例5
1-(2,3,4-三甲氧苄基)-3-氧代-1,2,3,4-四氢喹喔啉-6-甲酰正丁胺(7a)的制备
将化合物6(25mg,0.0671mmol),甲苯(3mL),草酰氯(57μL,0.671mmol,10euqiv)依次投入到25mL茄型瓶中,室温搅拌至固体全部溶解,升温至70℃反应3小时,反应完毕,蒸出甲苯及未反应的草酰氯,残余物即为酰氯中间体,未纯化;将正丁胺(20μL,0.201mmol,3equiv),无水四氢呋喃(1.0mL),无水碳酸钾(28mg,0.201mmol,3equiv)依次投入到10mL茄型瓶中,用无水四氢呋喃(1.0mL)将上述制备的酰氯中间体溶解,慢慢滴加到反应液中,室温搅拌反应20分钟,反应完毕,蒸出溶剂,柱层析(200-300目硅胶,二氯甲烷︰甲醇=80:1),得到21mg白色固体,收率73.2%。熔点:184.5-186.0℃。1H NMR(300MHz,CDCl3)δ8.72(s,1H),7.52(s,1H),7.25(d,J=8.4,1H),6.72(d,J=8.5Hz,1H),6.48(s,2H),6.06(t,J=5.4Hz,1H),4.39(s,2H),3.90(s,2H),3.84(s,3H),3.81(s,6H),3.44-3.51(m,2H),1.62-1.55(m,2H),1.47-1.37(m,2H),0.95(t,J=7.3Hz,3H).13C NMR(75MHz,CDCl3)δ166.6,165.6,153.7,137.7,137.5,131.3,126.0,124.9,122.2,115.3,111.1,104.3,60.9,56.2,53.9,52.0,39.9,31.8,20.2,13.8.HRMS(ESI):calcd for C23H30N3O5[M+H]+428.21800,found428.21802.
实施例6
1-(2,3,4-三甲氧苄基)-3-氧代-1,2,3,4-四氢喹喔啉-6-甲酰环己胺(7b)的制备
除用环己胺代替正丁胺外,其他步骤及提纯方法同实施例5,得到19mg淡黄色固体,收率62.4%。熔点:167.1-169.0℃。1H NMR(300MHz,CDCl3)δ8.80(s,1H),7.55(d,J=1.6Hz,1H),7.24(d,J=8.1,1H),6.70(d,J=8.4Hz,1H),6.48(s,2H),5.91(d,J=7.9Hz,1H),4.39(s,2H),4.01-4.07(m,1H),3.90(s,2H),3.84(s,3H),3.81(s,6H),1.95-2.08(m,2H),1.68-1.80(m,3H),1.39-1.50(m,2H),1.15-1.23(m,3H).13C NMR(75MHz,CDCl3)δ165.7,165.6,153.7,137.6,137.5,131.3,126.1,125.1,122.1,115.5,111.0,104.3,60.9,56.2,53.9,52.1,48.7,33.4,25.6,24.9.HRMS(ESI):calcd for C25H32N3O5[M+H]+454.23365,found 454.23367.
实施例7
1-(4-氟苯甲酰基)-3-氧代-1,2,3,4-四氢喹喔啉-6-甲酸甲酯(10a)的制备
(1)4-氟苯甲酰氯的制备(9a)
将对氟苯甲酸8a(110mg,0.786mmol,2equiv)、氯化亚砜(856μL,11.8mmol,30equiv)依次投入到10mL茄型瓶中,升温至70℃反应3h,反应完毕,减压蒸除氯化亚砜,得到淡黄色油状物,未纯化;
(2)将4a(81mg,0.393mmol),无水四氢呋喃(1.0mL),无水碳酸钾(109mg,0.786mmol,2equiv)依次投入到10mL茄型瓶中,用无水四氢呋喃(1.0mL)将上述制备化合物9a溶解,慢慢滴加至茄型瓶中,室温下搅拌反应2小时,减压蒸除溶剂,用200-300目硅胶柱层析(洗脱剂DCM:MeOH=80︰1),得到77mg灰白色固体,收率59.7%。熔点:239.6-241.5℃。1H NMR(300MHz,CDCl3)δ9.31(s,1H),7.71(d,J=1.2Hz,1H),7.39-7.53(m,3H),7.02(t,J=8.5Hz,2H),6.70(d,J=8.5Hz,1H),4.60(s,2H),3.91(s,3H).13C NMR(75MHz,CDCl3)δ168.2,167.9,165.9,164.5(J=252),131.6(J=8.25),131.5,129.9,129.6(J=3.75),127.8,124.3,124.2,117.9,115.9(J=21.7),52.5,47.6.HRMS(ESI):calcd for C17H14FN2O4[M+H]+329.09321,found 329.09311.
实施例8
1-(4-甲氧基苯甲酰基)-3-氧代-1,2,3,4-四氢喹喔啉-6-甲酸甲酯(10b)的制备
除步骤(1)中用对甲氧基苯甲酸替代对氟苯甲酸制得化合物9b外,其他步骤及提纯方法同实施例7,得到78mg灰白色固体,收率58.3%。熔点:205.5-207.2℃。1H NMR(300MHz,CDCl3)δ9.37(s,1H),7.70(d,J=1.7Hz,1H),7.46(dd,J=8.5,1.8Hz,1H),7.39(d,J=8.8Hz,2H),6.81(d,J=8.8Hz,2H),6.73(d,J=8.5Hz,1H),4.59(s,2H),3.90(s,3H),3.82(s,3H).13C NMR(75MHz,CDCl3)δ169.0,168.2,166.0,162.3,132.2,131.3,129.7,127.3,125.4,124.3,124.2,117.8,113.8,55.4,52.5,47.7.HRMS(ESI):calcd for C18H17N2O5[M+H]+341.11320,found341.11310.
实施例9
1-(3,4-二甲氧基苯甲酰基)-3-氧代-1,2,3,4-四氢喹喔啉-6-甲酸甲酯(10c)的制备
除步骤(1)中用3,4-二甲氧基苯甲酸替代对氟苯甲酸制得化合物9c外,其他步骤及提纯方法同实施例7,得到68mg白色固体,收率46.7%。熔点:217.0-218.8℃。1H NMR(300MHz,CDCl3)δ9.39(s,1H),7.70(d,J=1.7Hz,1H),7.47(dd,J=8.5,1.8Hz,1H),7.04(d,J=1.9Hz,1H),6.95(dd,J=8.3,1.9Hz,1H),6.74(m,2H),4.59(s,2H),3.90(s,3H),3.88(s,3H),3.78(s,3H).13C NMR(75MHz,CDCl3)δ169.0,168.1,166.0,152.0,148.9,132.2,129.6,127.3,125.4,124.3,124.2,123.1,117.8,112.2,110.3,56.0,52.5,47.8.HRMS(ESI):calcd for C19H19N2O6[M+H]+371.12376,found 371.12363.
实施例10
1-(3,4,5-三甲氧基苯甲酰基)-3-氧代-1,2,3,4-四氢喹喔啉-6-甲酸甲酯(10d)的制备
除步骤(1)中用3,4,5-三甲氧基苯甲酸替代对氟苯甲酸制得化合物9d外,其他步骤及提纯方法同实施例7,得到92mg灰白色固体,收率58.5%。熔点:92.0-94.6℃。1H NMR(300MHz,CDCl3)δ8.99(brs,1H),7.67(s,1H),7.51(d,J=8.2Hz,1H),6.83(d,J=8.2Hz,1H),6.64(s,2H),4.59(s,2H),3.91(s,3H),3.87(s,3H),3.70(s,6H).13C NMR(75MHz,CDCl3)δ168.9,167.7,165.9,153.1,141.0,131.7,129.6,128.2,127.5,124.3(2C),117.6,106.5,61.1,56.2,52.5,47.8.HRMS(ESI):calcd for C20H21N2O7[M+H]+401.13433,found 401.13437.
实施例11
1-[2-(3,4,5)-三甲氧基乙酰基]-3-氧代-1,2,3,4-四氢喹喔啉-6-甲酸甲酯(10e)的制备
除步骤(1)中用3,4,5-三甲氧基苯乙酸替代对氟苯甲酸外,其他步骤及提纯方法同实施例7,得到103mg白色固体,收率63.2%。熔点:188.0-181.9℃。1H NMR(300MHz,CDCl3)δ9.10(s,1H),7.77(dd,J=8.3,1.3Hz,1H),7.61(s,1H),7.41-7.52(m,1H),6.35(s,2H),4.51(s,2H),3.93(s,3H),3.85(d,J=4.5Hz,2H),3.80(s,3H),3.78(s,6H).13C NMR(75MHz,CDCl3)δ170.4,165.8,153.4,137.0,130.8,129.3,128.7,124.6,124.2(2C),117.9,106.4,105.6,60.9,56.1,52.6,41.4,41.1.HRMS(ESI):calcd for C21H23N2O7[M+H]+415.14998,found 415.14999.
实施例12
1-(3,4-二甲氧基苯甲酰基)-3-氧代-1,2,3,4-四氢喹喔啉-6-甲酰丁胺(10f)的制备
(1)3-硝基-4-氨基苯甲酰丁胺的制备(2b)
将3-硝基-4-氨基苯甲酸(250mg,1.37mmol),二氯甲烷(30mL),EDCI(314mg,1.64mmol,1.2equiv),正丁胺(135μL,1.37mmol,1equiv)依次投入到100mL茄型瓶中,室温搅拌反应2小时,反应完毕,蒸出溶剂,柱层析(200-300目硅胶,石油醚︰乙酸乙酯=1︰1),得到273mg黄色固体,收率84.0%。熔点:162.8-164.6℃。1H NMR(300MHz,CDCl3)δ8.47(s,1H),7.90(d,J=8.8Hz,1H),6.86(d,J=8.7Hz,1H),6.35(brs,2H),6.05(brs,1H),3.42-3.49(m,2H),1.58-1.67(m,2H),1.35-1.49(m,2H),0.97(t,J=7.3Hz,3H).MS(ESI)[M+H]+238.08。
(2)3-硝基-4-(2-乙氧基-2-氧代乙基)胺基苯甲酰丁胺的制备(3b)
将2b(200mg,0.843mmol),碳酸铯(329mg,1.01mmol,1.2equiv),溴乙酸乙酯(0.935mL,8.43mmol,10equiv)依次投入到25mL圆底烧瓶中,升温至140℃反应20小时,反应完毕,减压蒸出溴乙酸乙酯,残留物经柱层析(石油醚:乙酸乙酯=20︰1),得126mg黄色固体,收率46.2%。熔点:152.8-154.0℃。1H NMR(300MHz,CDCl3)δ8.66(brs,1H),8.55(s,1H),8.01(d,J=8.6Hz,1H),6.76(d,J=9.0Hz,1H),6.05(brs,1H),4.31(q,J=7.5Hz,2H),4.13(d,J=5.1Hz,2H),3.40-3.51(m,2H),1.57-1.67(m,2H),1.37-1.49(m,2H),1.33(t,J=7.1Hz,3H),0.97(t,J=7.3Hz,3H).MS(ESI)[M-1]-322.41.
(3)3-氧代-1,2,3,4-四氢喹喔啉-6-甲酸甲酯的制备(4b)
将3b(300mg,0.928mmol),无水甲醇(100mL),10%钯碳(49mg,0.0464mmol,0.05equiv)依次投入到250mL圆底烧瓶中,氢气球置换空气三次,室温搅拌反应10小时,反应完毕,抽滤,蒸出溶剂,柱层析(200-300目硅胶,二氯甲烷︰甲醇=80:1),得到158mg灰白色固体,收率68.8%。熔点:211.9-213.3℃。1H NMR(300MHz,CD3OD)δ7.32(d,J=8.3Hz,1H),7.24(s,1H),6.67(d,J=8.3Hz,1H),3.92(s,2H),3.32-3.37(m,2H),1.54-1.63(m,2H),1.35-1.44(m,2H),0.96(t,J=7.2Hz,3H).MS(ESI)[M-1]-246.44.
(4)1-(3,4-二甲氧基苯甲酰基)-3-氧代-1,2,3,4-四氢喹喔啉-6-甲酰丁胺的制备(10f)
将4b(60mg,0.243mmol)、无水四氢呋喃(1.0mL)、无水碳酸钾(67mg,0.486mmol,2equiv)依次投入到10mL茄型瓶中,用无水四氢呋喃(1.0mL)将化合物9c(2equiv)溶解,慢慢滴加至茄型瓶中,室温下搅拌反应2小时,减压蒸除溶剂,用200-300目硅胶柱层析(洗脱剂DCM:MeOH=80︰1),得到55mg白色固体,收率55.1%。熔点:224.5-226.7℃。1H NMR(300MHz,CD3OD)δ7.50(s,1H),7.20(d,J=8.5Hz,1H),6.95-7.02(m,2H),6.89(d,J=8.7Hz,1H),6.79(d,J=8.5Hz,1H),4.50(s,2H),3.82(s,3H),3.69(s,3H),3.31-3.39(m,2H),1.51-1.63(m,2H),1.34-1.42(m,2H),0.95(t,J=7.2Hz,3H).13C NMR(75MHz,CD3OD)δ169.5,168.1,167.4,152.0,148.8,132.1,130.9,130.4,125.8,124.0,122.7,120.5,115.6,112.2,110.6,55.0(2C),39.4,31.2,19.8,12.8.HRMS(ESI):calcd for C22H26N3O5[M+H]+412.18670,found 412.18668.
实施例13
1-(3,4,5-三甲氧基苯甲酰基)-3-氧代-1,2,3,4-四氢喹喔啉-6-甲酰丁胺(10g)的制备
除步骤(4)中用9d替代9c外,其他步骤及提纯方法同实施例12,得到70mg灰白色固体,收率65.3%。熔点:230.5-232.0℃。1H NMR(300MHz,CD3OD)δ7.50(s,1H),7.23(d,J=8.5Hz,1H),6.86(d,J=8.5Hz,1H),6.70(s,2H),4.51(s,2H),3.77(s,3H),3.69(s,6H),3.32-3.37(m,2H),1.52-1.62(m,2H),1.34-1.44(m,2H),0.95(t,J=7.3Hz,3H).13C NMR(75MHz,CD3OD)δ169.4,167.9,167.4,153.1,140.5,132.3,131.0,130.1,129.0,124.1,120.5,115.5,106.3,59.8,55.2,39.4,31.2,19.8,12.7.HRMS(ESI):calcd for C23H28N3O6[M+H]+442.19726,found 442.19727.
实施例14
1-(3,4,5-三甲氧基苯甲酰基)-3-氧代-1,2,3,4-四氢喹喔啉-6-甲酰环己胺(10h)的制备
除步骤(1)中用环己胺替代正丁胺、步骤(4)中用9d替代9c外,其他步骤及提纯方法同实施例12,得到62mg灰白色固体,收率54.6%。熔点:263.9-264.9℃。1H NMR(300MHz,CDCl3)δ9.07(s,1H),7.84(s,1H),7.07(d,J=8.4Hz,1H),6.83(d,J=8.5Hz,1H),6.66(s,2H),5.98(d,J=8.0Hz,1H),4.56(s,2H),4.01-4.10(m,1H),3.88(s,3H),3.73(s,6H),1.99-2.08(m,2H),1.64-1.80(m,3H),1.24-1.53(m,2H),1.25-1.29(m,3H).13C NMR(75MHz,CDCl3)δ169.0,167.0,164.9,153.2,140.8,132.0,130.6,130.2,128.6,124.0,119.8,116.8,106.4,61.0,56.3,49.1,48.1,33.2,25.5,24.8.HRMS(ESI):calcd for C25H30N3O6[M+H]+468.21291,found468.21289.
实施例15
1-(3,4-二甲氧基苯甲酰基)-3-氧代-1,2,3,4-四氢喹喔啉-6-甲酰苯胺(10i)的制备
除步骤(1)中用苯胺替代正丁胺,其他步骤及提纯方法同实施例12,得到54mg白色固体,收率51.5%。熔点:274.7-276.5℃。1H NMR(300MHz,CDCl3)δ8.25(s,1H),7.75(s,1H),7.56-7.65(m,3H),7.38(t,J=7.5Hz,2H),7.22-7.25(m,1H),7.17(t,J=7.0Hz,1H),7.07(s,1H),6.97(d,J=8.8Hz,1H),6.85(d,J=8.4Hz,1H),6.76(d,J=8.3Hz,1H),4.58(s,2H),3.90(s,3H),3.82(s,3H).13C NMR(75MHz,CDCl3)δ167.2,164.0(2C),152.0,149.0,137.5,131.2,129.2,125.5(2C),125.0(2C),124.4,123.0,120.6,120.2,116.0,112.1,110.3,56.0,48.0.HRMS(ESI):calcd for C24H22N3O5[M+H]+432.15540,found 432.15540.
实施例16
1-(3,4-三甲氧基苯甲酰基)-3-氧代-1,2,3,4-四氢喹喔啉-6-甲酰苯胺(10j)的制备
除步骤(1)中用苯胺替代正丁胺、步骤(4)中用9d替代9c外,其他步骤及提纯方法同实施例12,得到62mg白色固体,收率55.3%。熔点:247.1-248.5℃。1H NMR(300MHz,CDCl3)δ9.11(s,1H),8.07(s,1H),7.61-7.66(m,3H),7.36(t,J=7.4Hz,2H),7.27-7.30(m,1H),7.15(t,J=7.0Hz,1H),6.93(d,J=8.3Hz,1H),6.67(s,2H),4.53(s,2H),3.88(s,3H),3.73(s,6H).13C NMR(75MHz,CDCl3)δ169.2,167.3,164.2,153.2,140.9,137.6,132.3,130.5,130.4,129.2,128.4,125.0,124.3,120.8,120.4,116.3,106.4,61.0,56.3,48.2.HRMS(ESI):calcd for C25H24N3O6[M+H]+462.16596,found 462.16595.
实施例17
1-(3,4-二甲氧基苯甲酰基)-3-氧代-1,2,3,4-四氢喹喔啉-6-甲酰吗啉胺(10k)的制备
除步骤(1)中用吗啉替代正丁胺,其他步骤及提纯方法同实施例12,得到54mg淡黄色固体,收率50.3%。熔点:204.0-205.1℃。1H NMR(300MHz,CDCl3)δ8.62(s,1H),7.11(d,J=13.8Hz,2H),6.94(d,J=8.6Hz,1H),6.83(s,2H),6.74(d,J=8.1Hz,1H),4.55(s,2H),3.89(s,3H),3.83(s,3H),3.69(brs,4H),3.54-3.58(m,4H).13C NMR(75MHz,CDCl3)δ169.0(2C),167.5,151.9,149.0,132.5,130.2,129.6,125.5,124.2,122.9,121.6,116.1,112.1,110.3,66.8,56.0(2C),48.1.HRMS(ESI):calcd for C22H24N3O6[M+H]+426.16596,found 426.16595.
实施例18
1-(3,4,5-三甲氧基苯甲酰基)-3-氧代-1,2,3,4-四氢喹喔啉-6-甲酰吗啉胺(10l)的制备
除步骤(1)中用吗啉替代正丁胺、步骤(4)中用9d替代9c外,其他步骤及提纯方法同实施例12,得到59mg淡黄色固体,收率53.3%。熔点:205.5-207.0℃。1H NMR(300MHz,CDCl3)δ8.86(s,1H),7.16(s,1H),6.94(d,J=8.1Hz,1H),6.88(d,J=8.3Hz,1H),6.66(s,2H),4.54(s,2H),3.87(s,3H),3.74(s,6H),3.69(brs,4H),3.50-3.61(m,4H).13C NMR(75MHz,CDCl3)δ169.0,168.8,167.4,153.2,141.0,132.8,130.4,129.0,128.4,124.3,121.5,116.1,106.4,66.8,61.0,56.3,48.3.HRMS(ESI):calcd for C23H26N3O7[M+H]+456.17653,found 456.17651.
下面是本发明所述的部分药理实验及结果:
实验例1:对肿瘤细胞的增殖抑制实验(MTT实验)
(1)实验方法
测试本发明所述化合物对人类肝癌细胞株(SMMC-7721),人类子宫颈癌细胞株(HeLa),人类白血病细胞株(K562)的细胞增殖抑制活性。以上细胞株均由该实验室冻存和传代。方法:对于SMMC-7721和HeLa两种肿瘤细胞,将细胞接种于96孔平底板中(每孔约有4000-6000个细胞),于37℃,5%CO2条件下孵育24小时后,每孔加入不同浓度待测化合物或阳性对照药,再培养72小时后,各小孔中分别加入50μL MTT的PBS缓冲液(浓度为1mg/mL),与37℃孵育4小时,除去MTT和培养基,每个小孔中分别加入100μL DMSO,于570nm下检测(Power Wave XS,Bio Tek,美国);对于K562细胞株,将细胞接种于96孔平底板中(每孔约有6000个细胞),与37℃,5%CO2条件下孵育24小时后,每孔加入不同浓度待测化合物或阳性对照药,再培养72小时后,各小孔中分别加入50μL MTT的无血清培养基溶液(浓度为1mg/mL),与37℃孵育4小时,每小孔中加入50μL三联溶液(10%SDS,5%异丁醇,0.01mol/L盐酸),37℃孵育12小时,待温度降至室温于570nm下检测(Power Wave XS,Bio Tek,美国)。选取康普瑞汀(CA-4)、阿霉素为阳性对照药。对数据进行统计分析。
(2)实验结果
实验结果如表1所示。结果显示该类化合物普遍具有较强的细胞生长抑制作用。化合物5c、10c对细胞生长抑制作用的半数抑制浓度(IC50)达到几微摩尔/升(μM),化合物10d达到纳摩尔水平(nM)。这些化合物的细胞生长抑制活性明显优于阿霉素。
表1化合物MTT测试结果
说明:表格中化合物CA-4,阿霉素为阳性对照药,不包含在专利保护范围。
实验例2:微管蛋白聚合抑制实验
(1)实验方法
测试本发明所述化合物对微管蛋白聚合抑制作用,采用cytoskeleton公司BK011P试剂盒。将96孔板置于多功能酶标仪(CLARIOstar,BMG LABTECH公司,德国)上,37℃预热10分钟。按照说明书上操作,配置好紫杉醇溶液(10×)、CA-4溶液(10×)、检测化合物溶液(10×)、缓冲液1、GTP母液、微管蛋白甘油缓冲液、微管蛋白母液备用,根据实验选择合适的比例,将缓冲液1、GTP母液、微管蛋白甘油缓冲液、微管蛋白母液混合配制微管反应液,置于冰浴上。向96孔板中分别加入5μL对照缓冲液、紫杉醇溶液、CA-4溶液、待测化合物溶液(设1复孔),酶标仪上37℃预热1分钟,快速加入上面配制好的微管反应液,记录60分钟内荧光强度变化。
缓冲液1的配制:①每瓶缓冲液1用10mL Milli-Q超纯水溶解其中的物质(共两瓶);②混合两瓶中的物质;③将上述缓冲液分装到13个冻存管中,每管装1.5mL,-70℃条件下保存。
微管蛋白甘油缓冲液,保存于4℃。
GTP母液的配制:①每管GTP母液用100μL的冰冷无菌蒸馏水溶解其中的物质(共3管);②将其置于冰上;③混合三管中物质;④将上述溶液分装到13个冻存管中,每管20μL,-70℃条件下保存。
微管蛋白溶液的配制:①将盛装冻干微管蛋白的小瓶放在冰上,将液氮置于杜瓦瓶中;②标记12个冻存管并置于冰上待用;③将一个20μLGTP母液除霜;④将1.5mL冰冷的缓冲液1与15μLGTP母液混合;⑤用1.1mL上述混合液溶解微管蛋白冻干粉末;⑥置于冰上2分钟待粉末全部溶解悬浮;⑦将上述微管蛋白液分装到12个细胞冻存管中,每管88μL,置于液氮中迅速冷冻;⑧-70℃条件下保存。
紫杉醇母液配制:100μL DMSO溶解紫杉醇,-70℃条件下保存。
(2)实验结果
结果显示化合物5c、10c及10d在20μM具有明显的抑制微管蛋白聚合抑制作用,化合物10d对微管蛋白聚合抑制作用的半数抑制浓度(IC50)为3.9μM,如附图1所示。
实验例3:细胞周期实验
(1)实验方法
测试本发明所述化合物对癌细胞周期作用(选取人类子宫颈癌细胞株HeLa),采用PI(碘化丙啶)染色。方法:将细胞接种于6孔平底板中(每孔约有100000个细胞),于37℃,5%CO2条件下孵育24小时后,每孔加入不同浓度10d(0.10μM,0.15μM,0.20μM,0.25μM),设置空白对照组(空白对照组不加化合物10d),于37℃,5%CO2条件下孵育24小时。收集细胞,用预冷PBS(4℃,0.01mol/L)洗涤,将细胞加入到预冷(4℃)的70%(v/v)乙醇中,固定好的细胞-20℃条件下放置过夜。离心,除去乙醇,预冷PBS(4℃,0.01mol/L)洗涤,加入带有RNase的PI染料于4℃条件下避光染色1小时,离心,除去染料,PBS洗涤,过300目滤网,在流式细胞仪(BD FACSVerse,美国)上进行测试。
(2)实验结果
结果显示化合物10d在0.15μM能够明显地将细胞阻滞在G2/M生长期。
- 还没有人留言评论。精彩留言会获得点赞!