一种新的羊毛甾烷型三萜化合物及其制备方法和医药用途与流程
本发明属于药物
技术领域:
,具体涉及从干燥的野马追中分离得到的一种具有治疗骨破坏性疾病作用的羊毛甾烷型三萜类化合物及其制备方法。
背景技术:
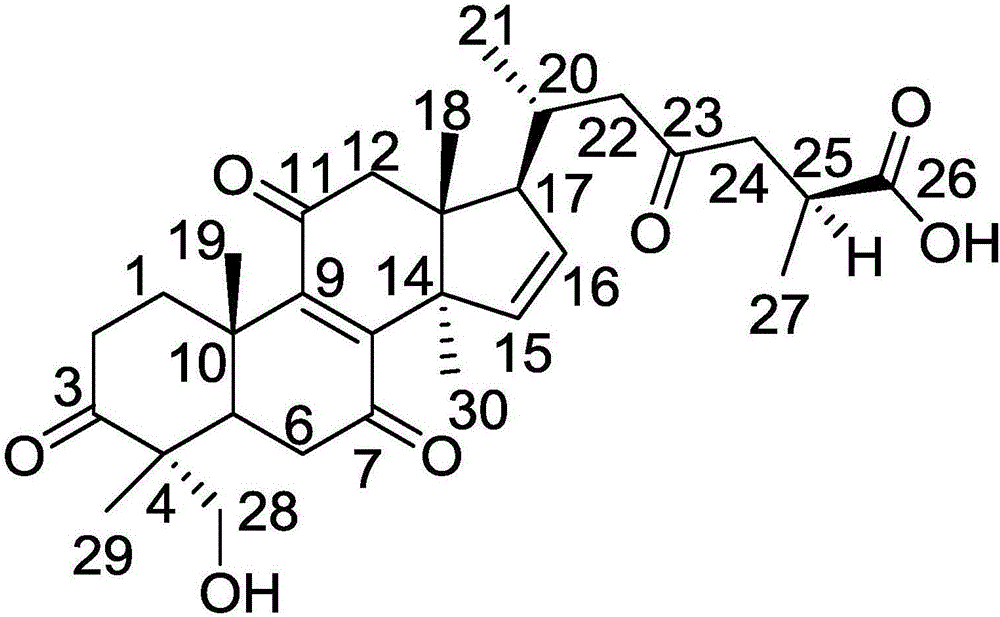
:野马追是菊科植物轮叶泽兰EupatoriumlindleyanumDC.的干燥地上部分,又名白鼓钉(《江苏南部种子植物手册》)、化食草(《杭州药用植物志》)、毛泽兰(《内蒙古植物志》),产于江苏、甘肃、山东、湖南等地。其味苦,性平,归肝、脾经,具有化痰止咳、清热解毒、利尿消肿、降压的功能,用于治疗感冒、咳嗽多痰、头疼、扁桃体炎、细菌性痢疾、高血压、慢性气管炎、支气管炎等。其枝叶入药有解表祛湿、和中化湿之效,用于劳伤咳嗽、吐血咳血以及淋浊白带、无名肿痛等证的治疗。迄今为止,从野马追植物中发现的化学成分类型主要包括倍半萜类、黄酮类、三萜类、挥发油类、有机酸类及其他类成分,其中以倍半萜类化合物的研究最多。迄今为止,已得到的倍半萜类化合物主要为愈创木烷型和吉马烷型,此外还有少量的杜松烷型和桉烷型倍半萜类。药理作用研究表明,野马追提取物具有抗炎、抗菌、降血脂以及保护急性肺损伤、止咳、平喘等作用。野马追广泛应用于临床,主要包括片剂、颗粒剂、糖浆剂等制剂品种,还有以野马追为君药的复方野马追颗粒、胶囊等产品,主要用于治疗慢性气管炎、支气管炎、高血压等症。通过对临床应用的观察发现,野马追注射液治疗钩端螺旋体病具有退热显著、缓解症状快、治愈时间短的特点。同时,野马追糖浆用于治疗小儿咳喘性疾病也安全、有效。技术实现要素:本发明的目的是提供一种从干燥的野马追中分离得到的一种具有治疗骨破坏性疾病作用的羊毛甾烷型三萜类化合物及其制备方法。本发明的上述目的是通过下面的技术方案得以实现的:具有下述结构式的化合物(Ⅰ),所述的化合物(Ⅰ)的制备方法,包含以下操作步骤:(a)将干燥的野马追粉碎,用80~90%乙醇热回流提取,合并提取液,浓缩至无醇味,依次用石油醚、乙酸乙酯和水饱和的正丁醇萃取,分别得到石油醚萃取物、乙酸乙酯萃取物和正丁醇萃取物;(b)步骤(a)中乙酸乙酯萃取物用大孔树脂除杂,先用10%乙醇洗脱6个柱体积,再用80%乙醇洗脱8个柱体积,收集80%乙醇洗脱液,减压浓缩得80%乙醇洗脱物浸膏;(c)步骤(b)中80%乙醇洗脱浸膏用正相硅胶分离,依次用体积比为80:1、50:1、35:1、15:1和1:1的二氯甲烷-甲醇梯度洗脱得到5个组分;(d)步骤(c)中组分4用正相硅胶进一步分离,依次用体积比为25:1、15:1和5:1的二氯甲烷-甲醇梯度洗脱得到3个组分;(e)步骤(d)中组分2用十八烷基硅烷键合的反相硅胶分离,用体积百分浓度为75%的甲醇水溶液等度洗脱,收集8~10个柱体积洗脱液,洗脱液减压浓缩得到纯的化合物(Ⅰ)。进一步地,所述大孔树脂为AB-8型大孔吸附树脂。进一步地,所述用乙醇热回流提取采用的乙醇浓度为85%。一种药物组合物,其中含有治疗有效量的所述的化合物(Ⅰ)和药学上可接受的载体。所述的化合物(Ⅰ)在制备治疗骨破坏性疾病的药物中的应用。所述的药物组合物在制备治疗骨破坏性疾病的药物中的应用。本发明化合物用作药物时,可以直接使用,或者以药物组合物的形式使用。该药物组合物含有治疗有效量的本发明化合物(Ⅰ),其余为药物学上可接受的、对人和动物无毒和惰性的可药用载体和/或赋形剂。所述的可药用载体或赋形剂是一种或多种选自固体、半固体和液体稀释剂、填料以及药物制品辅剂。将本发明的药物组合物以单位体重服用量的形式使用。本发明药物可通过口服或注射的形式施用于需要治疗的患者。用于口服时,可将其制成片剂、缓释片、控释片、胶囊、滴丸、微丸、混悬剂、乳剂、散剂或颗粒剂、口服液等;用于注射时,可制成灭菌的水性或油性溶液、无菌粉针、脂质体或乳剂等。附图说明图1为化合物(Ⅰ)结构式;图2为化合物(Ⅰ)理论ECD值与实验ECD值比较。具体实施方式下面结合实施例进一步说明本发明的实质性内容,但并不以此限定本发明保护范围。尽管参照较佳实施例对本发明作了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的实质和范围。实施例1:化合物(Ⅰ)分离制备及结构确证试剂来源:乙醇、石油醚、乙酸乙酯、正丁醇、二氯甲烷为分析纯,购自上海凌峰化学试剂有限公司,甲醇,分析纯,购自江苏汉邦化学试剂有限公司。制备方法:(a)将干燥的野马追(8kg)粉碎,用85%乙醇热回流提取(25L×3次),合并提取液,浓缩至无醇味(3L),依次用石油醚(3L×3次)、乙酸乙酯(3L×3次)和水饱和的正丁醇(3L×3次)萃取,分别得到石油醚萃取物、乙酸乙酯萃取物(359g)和正丁醇萃取物;(b)步骤(a)中乙酸乙酯萃取物用AB-8型大孔树脂除杂,先用10%乙醇洗脱6个柱体积,再用80%乙醇洗脱8个柱体积,收集80%乙醇洗脱液,减压浓缩得80%乙醇洗脱物浸膏(138g);(c)步骤(b)中80%乙醇洗脱浸膏用正相硅胶分离,依次用体积比为80:1(8个柱体积)、50:1(8个柱体积)、35:1(6个柱体积)、15:1(8个柱体积)和1:1(5个柱体积)的二氯甲烷-甲醇梯度洗脱得到5个组分;(d)步骤(c)中组分4(32g)用正相硅胶进一步分离,依次用体积比为25:1(8个柱体积)、15:1(10个柱体积)和5:1(6个柱体积)的二氯甲烷-甲醇梯度洗脱得到3个组分;(e)步骤(d)中组分2(17g)用十八烷基硅烷键合的反相硅胶分离,用体积百分浓度为75%的甲醇水溶液等度洗脱,收集8-10个柱体积洗脱液,洗脱液减压浓缩得到纯的化合物(Ⅰ)(29mg)。结构确证:白色针状结晶;HR-ESIMS显示[M+Na]+为m/z535.2712,结合核磁特征可得分子式为C30H40O7,不饱和度为11。核磁共振氢谱数据δH(ppm,DMSO-d6,600MHz):H-1(1.57,m),H-1(1.85,m),H-2(2.21,m),H-2(2.37,m),H-5(1.79,m),H-6(2.74,m),H-6(3.02,m),H-12(2.71,m),H-12(2.89,m),H-15(5.63,m),H-16(6.25,m),H-17(1.90,m),H-18(1.11,s),H-19(1.26,s),H-20(1.78,m),H-21(0.98,d,J=6.3),H-22(2.27,m),H-22(2.61,m),H-24(2.52,m),H-24(3.15,m),H-25(3.24,m),H-27(1.26,d,J=7.2),H-28(3.61,d,J=10.5),H-28(3.78,d,J=10.5),H-29(1.17,s),H-30(1.21,s);核磁共振碳谱数据δC(ppm,DMSO-d6,150MHz):37.1(CH2,1-C),34.3(CH2,2-C),215.9(C,3-C),52.5(C,4-C),43.8(CH,5-C),37.7(CH2,6-C),202.6(C,7-C),151.4(C,8-C),149.2(C,9-C),38.7(C,10-C),201.8(C,11-C),52.5(CH2,12-C),46.2(C,13-C),57.1(C,14-C),135.2(CH,15-C),133.8(CH,16-C),50.5(CH,17-C),18.7(CH3,18-C),18.5(CH3,19-C),33.1(CH,20-C),19.8(CH3,21-C),49.8(CH2,22-C),209.1(C,23-C),47.0(CH2,24-C),35.3(CH,25-C),178.7(C,26-C),18.1(CH3,27-C),68.2(CH2,28-C),17.4(CH3,29-C),24.1(CH3,30-C);碳原子标记参见图1。IR谱表明该化合物含有羟基(3425cm-1)和α,β-不饱和羰基(1697cm-1)基团。此外,化合物在268和226nm处有最大紫外吸收。1HNMR谱显示四个单峰甲基信号δH1.11,1.17,1.21和1.26,两个双峰甲基信号δH0.98(d,J=6.3Hz,H-21)和1.26(d,J=7.2Hz,H-27),一个含氧亚甲基信号δH3.61(d,J=10.5Hz,H-28)和3.78(d,J=10.5Hz,H-28),以及两个烯属次甲基信号δH5.63(m,H-15)和6.25(m,H-16)。13CNMR谱显示30个共振碳信号,六个甲基,七个亚甲基(一个含氧亚甲基),六个次甲基(两个烯属次甲基),十二个季碳(五个羰基碳,两个烯烃季碳)。其中,共振信号δC202.6(C-7),151.4(C-8),149.2(C-9)和201.8(C-11)为共轭系统(C-7/C-8/C-9/C-11)的标志。HMBC谱中H2-1,H2-2,H-5,H2-28和Me-29与C-3,Me-29和H-5与C-28的相关性,以及ROESY谱中H2-28和H-5的相关性表明C-3为酮羰基,C-28为羟基亚甲基。两个烯属次甲基质子信号[δH5.63(m,H-15)和6.25(m,H-16)]和碳信号[135.2(C-15),133.8(C-16)]表明C-15和C-16之间为双键。HMBC谱中Me-21与C-17,C-20和C-22,H-20与C-22和C-23,H2-22与C-23和C-24,H-25与C-24和C-26,以及Me-27与C-24,C-25和C-26的相关性表明C-23为酮羰基,C-26为羧基基团。综合氢谱、碳谱、HMBC谱和ROESY谱,以及文献关于相关类型核磁数据,可基本确定该化合物如图1所示,立体构型进一步通过ECD试验确定,理论值与实验值基本一致(图2)。实施例2:化合物(Ⅰ)药理作用试验一、材料和仪器SD雄性大鼠(4周龄)购于河北医科大学动物实验中心,清洁级,体重150g。化合物(Ⅰ)自制,HPLC归一化纯度大于98%。1α,25(OH)2D3购于英国普利茅斯生物研究实验室。α-MEM培养基购于美国GIBCOBRL。SolubleRANKL购于英国PeprotecScience。抗酒石酸酸性磷酸酶(TRAP)试剂盒购于美国Sigma公司。葡聚糖凝胶购于上海如吉科技发展有限公司。标准胎牛血清购于山东银香伟业生物有限公司。注射用青霉素钠购于华北制药有限公司。注射用硫酸链霉素购于深圳华药南方制药有限公司。丙酮、甲醛溶液购于石家庄市有机化工厂。异氟烷购于鲁南贝特制药有限公司。二甲基亚砜购于天津市光复精细化工研究所。BB5060/BB16二氧化碳培养箱(上海Heraeus公司),VD-650型桌上式细胞培养超净操作台(苏州净化设备有限公司),BFX5-320型低速离心机(中国上海泸南科学仪器联营厂),XD-1019810倒置显微镜(江南公司),CX-21光学显微镜(日本OLYMPUS),微量加样器(法国GILSON),2510型变频振荡仪(上海新波生物技术有限公司),GF-300型电子天(JapanA&DCompany,limited),725型超低温冰箱(FormaScientific.Inc)。未提到的材料和仪器均为常规仪器。未提到的溶液配制均采用本领域常规溶液配制方法配制即可。药液配制及葡聚糖凝胶柱的制备:1、α-MEM培养液的制备:(1)α-MEM粉一袋+超纯水800ml,完全溶解;(2)加碳酸氢钠3.02g待完全溶解后加盐酸测PH达7.2;(3)再次加超纯净水至1000ml;(4)加抗生素(青霉素及链霉素);(5)滤过灭菌(0.22微米微孔滤膜过滤)(6)500ml灭菌瓶分装,4℃储藏备用。2、1α,25(OH)2D3储存液的分装(1)取1α,25(OH)2D3原液50微升(50微升一支),加入无水乙醇1150微升,使总量至1200微升(即24倍稀释)(2)以100微升/只分装于12支灭菌试管内,-30℃冰箱内储存备用;(3)应用前再次稀释1000倍,使最终浓度为1×10-8M。3、SolubleRANKL储存液分装(1)取RANKL冻干粉一支(10微克);(2)0.1Mtris-HClbuffer50微升溶解RANKL;(3)以5微升/只分装于10支灭菌冷冻管内,-30℃冰箱内储存备用;(4)使用前取出一支,先用含10%FBSα-MEM培养液495微升溶解(即稀释100倍);添加前再次稀释100倍,使终浓度为20ng/ml。4、0.1Mtris-HClbuffer的制备(1)tris-HCl12.11g(0.1M);(2)超纯水800ml(添加溶解后,调节pH至8.2);(3)加超纯水至1000ml备用。5、化合物(Ⅰ)储存液的制备(1)储存液的浓度:1mg/ml及100ug/ml。(2)储存液的配制:称取化合物(Ⅰ)1mg放于灭菌冷冻管内,加入二甲基亚砜1ml完全溶解,配成1mg/ml的储存液。从1mg/ml的储存液中取出100微升放于另一支灭菌冷冻管内,加入二甲基亚砜900微升即配成100微克/毫升的储存液,储存于4℃恒温冰箱,使用时间不超过两周。1mg/ml的储存液冷冻保存。6、TRAP固定液的制备在染色之前,按产品说明书配制。首先取无菌20ml试管一支,按以下顺序依次加入试剂:丙酮6.5ml、枸橼酸2.5ml、甲醛0.8ml,充分混匀,计9.8ml,现配现用。7、TRAP染色液的制备同样是在染色之前配制,按产品说明制备,现配现用。8、葡聚糖凝胶柱的制备(1)消毒后50ml注射器一个,除去内芯;(2)注射筒与注射头交接处置消毒后的脱脂棉球一个;(3)固定架固定针筒,针头处接关闭状的三通;(4)将灭菌的葡聚糖凝胶混匀,抽取30ml注入针筒内,打开三通,缓慢过滤沉淀,待凝胶沉淀液面至15ml时为止;关闭三通,放入4℃恒温冰箱过夜;(5)实验前1小时用含有15%胎牛血清的α-MEM培养液50ml缓慢注入凝胶柱内,打开三通,自然过滤洗涤凝胶柱后待用。二、试验方法1、全骨髓细胞培养1.1全骨髓细胞提取实验开始前30分钟将标准胎牛血清放入37℃水浴箱内解冻;取50mL无菌离心管一个,装入40mL不含胎牛血清的α-MEM培养液,以备冲洗全骨髓细胞用。(1)选4周龄SD雄性大鼠1只,75%乙醇消毒后,采用异氟烷吸入麻醉;(2)待麻醉起效后,无菌条件下取大鼠两侧胫骨和股骨,剪掉骨垢端暴露骨髓腔;(3)用10mL注射器抽取不含血清的α-MEM培养液,换用25号针头后自骨髓腔内冲出骨髓细胞至50mL无菌离心管内;(4)将所提取的细胞悬液充分吹打,待没有团块后,将盛有细胞的离心管与平衡管一起放入离心机内离心,调整转速(2000转/分),离心5分钟;离心过程中配制含有15%胎牛血清的α-MEM培养液50mL(7.5mL胎牛血清加入42.5mLα-MEM培养液中)备用;(5)离心结束后弃去上清液,加入含15%胎牛血清的α-MEM培养液20mL,充分吹打混匀,形成细胞悬液,此液简称为细胞原液。1.2细胞数的计算(原液浓度及稀释倍数)(1)取以上提取的细胞悬液25μL放入试管内,然后加入5%醋酸475μL,即形成20倍稀释的细胞悬液;将试管放在振荡器上,振荡20秒钟,目的是去除红细胞;(2)计算细胞数:取振荡后细胞悬液滴入细胞计数盘,勿超过边缘,盖玻片覆盖,倒置显微镜下计算细胞数,计数盘4个大格内所有细胞均计入。本实验计算细胞数为537个,根据公式(细胞数/4)×20×104cells/mL推算细胞原液的浓度,本实验计算如下(537/4)×20×104=26.85×106cells/mL即本实验细胞原液浓度为26.85×106cells/mL,而我们需要的目的浓度是2×106cells/mL,所以细胞原液需要稀释。(3)稀释倍数计算如下:稀释倍数=原液浓度/目的浓度;本实验稀释倍数=26.85×106cells/mL/2×106cells/mL=13.425,即原液需要稀释13.425倍才能达到需要的目的浓度。本实验需要2×106cells/mL的细胞悬液40mL,简称为目的量。(4)达到目的浓度所需原液的量,计算如下:达到目的浓度所需原液的量=目的量/稀释倍数。本实验计算如下:达到目的浓度所需原液的量=40mL/13.425≌2.98mL。即取26.85×106cells/mL的细胞原液2.98mL放入灭菌的50mL离心管内,然后加入37.02mL(40-2.98)含15%胎牛血清的α-MEM培养液即可配成2×106cells/mL的细胞悬液40mL,然后加入1α,25(OH)2D3储存液40μL,操作过程应避光,即1α,25(OH)2D3终浓度为1×10-8M;再加入4μLsolubleRANKL储存液,即solubleRANKL终浓度为20ng/mL;加液过程在超净工作台上操作,至此,所需2×106cells/mL的细胞悬液目的量配置完成。1.3培养细胞、加药(1)细胞接种:取4行6列24孔培养板一块,每孔加入2×106cells/mL的细胞悬液0.5mL(1×106cells)。(2)加入化合物(Ⅰ):取100μg/mL化合物(Ⅰ)分装液一支于培养板的第2列分别加入2.5μL,每加一孔均需换一个吸头;第3列每孔加入5μL;第4列每孔加入10μL。加完第4孔后,将此液放入4℃恒温冰箱保存,取1mg/mL化合物(Ⅰ)储存液一支。第5列每孔加入1mg/mL化合物(Ⅰ)储存液2.5μL;第6列每孔加入5μL。加完后剩余药液放入4℃恒温冰箱保存。第一列不加药物,作为对照组。至此在24孔培养板中化合物(Ⅰ)每行都有6个浓度,分别为0、0.5、1、2、5、10μg/mL,每个浓度每列4孔(n=4),第一列为对照组。(3)培养细胞:加完药后,将培养板放入CO2培养箱内,在37℃、5%CO2及饱和湿度条件下,培养7天。培养至第4天时换培养液一次,按上述方法配制含有15%胎牛血清的α-MEM培养液40m(l6mL胎牛血清加入34mLα-MEM培养液中),内含10μL青霉素8μL链霉素(80万u青霉素,100万链霉素分别用2mL灭菌蒸馏水溶解),加入4μLsolubleRANKL储存液,然后取1α,25(OH)2D3储存液40μL加入。从培养箱内取出培养板后,倒置显微镜下观察细胞生长状况,每孔吸出旧培养液300μL,加入新培养液400μL,每吸出一列更换一次吸头,每加一孔更换一次吸头。换液完成后,将培养板继续放入CO2培养箱内,在37℃、5%CO2及饱和湿度条件下,再培养3天。2、破骨前驱细胞(POC)培养2.1全骨髓细胞的提取:如同以上所述方法,制备全骨髓细胞悬液1.5mL。2.2非附着骨髓细胞的提取(1)把用培养液冲洗过的凝胶柱,固定在固定架上;(2)缓慢向凝胶柱内注入全骨髓细胞1.5mL,打开三通,缓慢过滤;待1.5mL全骨髓细胞渗入凝胶柱内后,再缓慢注入含15%胎牛血清的α-MEM培养液10mL。(3)收集含有非附着骨髓细胞的培养液共计12-15mL,此液称为非附着骨髓细胞原液。2.3细胞数的计算(原液浓度及稀释倍数)及达到目的浓度所需原液的量如同以上所述方法,计算细胞数,稀释倍数,达到目的浓度所需原液的量。本实验计算细胞数为337个,推算细胞原液的浓度为16.85×106cells/mL;稀释倍数为8.425,即细胞原液需稀释8.425倍才能达到需要的目的浓(2×106cells/mL);达到目的浓度所需原液的量40mL/8.425≌4.75mL,即取4.75mL细胞原液放入灭菌的50mL离心管内,然后加入35.25mL(40-4.75)含15%胎牛血清的α-MEM培养液即可配成2×106cells/mL的目的细胞悬液40mL,然后加入1α,25(OH)2D3储存液40ul,操作过程应避光,即1α,25(OH)2D3终浓度为1×10-8M;加入solubleRANKL储存液4uL,即solubleRANKL终浓度为20ng/mL;加液过程均在超净工作台上操作,至此,未附着骨髓细胞悬液目的量配置完成。2.4培养细胞及加药如同前述。3、倒置显微镜下观察3.1倒置显微镜的调节(1)将显微镜合轴;(2)将视野光圈开大至与聚光器光阑边缘一致;(3)将与物镜放大倍数一致的相板放入聚光器中;(4)把调中目镜换到目镜筒中,旋动调中目镜上的调焦环至相板相环的图像清晰;(5)调节聚光器上的相环调中旋钮,使两个圆环图像重叠成同心圆状态;(6)用普通目镜换下调中目镜。3.2TRAP染色(1)培养7天后,将培养板从培养箱内取出,弃去培养液;(2)加入固定液0.4mL/孔,固定1分钟;(3)弃去固定液,蒸馏水冲洗四次;(4)加入染色液3-4滴/孔,用锡纸包裹培养板;(5)在37℃、5%CO2及饱和湿度条件下孵育30分钟后,取出培养板,弃去染色液,蒸馏水冲洗四次,自然干燥。3.3观察培养7天后将培养板从培养箱内取出,弃去培养液之前,在倒置显微镜下观察不同药物浓度下细胞的生长状况及形态,决定是否染色。染色后带染色液倒置显微镜下观察染色效果。4、光镜观察带染色液倒置显微镜下观察后,弃去染色液,蒸馏水洗4次,自然干燥,普通光镜下观察计算破骨细胞数。5、统计学分析使用SPSS16.0统计学软件进行分析。数据处理采用mean±SD表示,对不同药物浓度相同处理时间,各组与对照组之间比较,化合物(Ⅰ)对破骨细胞形成及分化的影响,得出的数据资料分析采用方差分析,从而得出药效和药物浓度的变化关系。三、结果及结论在全骨髓细胞培养系中,0.5、1、2μg/mL组与对照组比较,TRAP染色阳性细胞(3核以上)数仅为对照组的49.04%、28.85%、5.77%,经统计学分析,加药组对破骨细胞的形成有抑制作用,当药物浓度为1μg/mL时与对照组相比,对破骨细胞形成有显著抑制作用(P﹤0.01)。见表1。在破骨前驱细胞培养系中,0.5、1、2、5μg/mL组与对照组相比,TRAP染色阳性细胞(单核或2核)数仅为对照组的85.11%、65.79%、37.83%、2.21%,经统计学分析,加药组对破骨前驱细胞的形成有抑制作用,当药物浓度为2μg/mL时与对照组相比,对破骨前驱细胞形成有显著抑制作用(P﹤0.01)。见表2。结论,本研究结果表明化合物(Ⅰ)对破骨细胞的分化及增殖具有抑制作用,且存在浓度依赖关系。应用化合物(Ⅰ)治疗以破骨细胞活性增强为主的骨破坏性疾病有可能成为一种有前景的治疗方法。表1全骨髓细胞培养系中TRAP染色阳性细胞(3核以上)数0(对照组)0.5μg/mL1μg/mL2μg/mL5μg/mL10μg/mL平行操作1835626520平行操作2814428400平行操作3812219400平行操作4673117502表2破骨前驱细胞培养系中TRAP染色阳性细胞(单核或2核)数实施例3片剂的制备:按实施例1方法先制得化合物(Ⅰ),以及利用有机酸如酒石酸、或柠檬酸或甲酸或乙二酸等、无机酸如盐酸或硫酸或磷酸制成的盐,按其与赋形剂重量比为1:9的比例加入赋形剂,制粒压片。实施例4口服液制备:按实施例1方法先制得化合物(Ⅰ),以及利用有机酸如酒石酸、或柠檬酸或甲酸或乙二酸等、无机酸如盐酸或硫酸或磷酸制成的盐,按常规口服液制法制成口服液。实施例5胶囊剂或颗粒剂的制备:按实施例1方法先制得化合物(Ⅰ),以及利用有机酸如酒石酸、或柠檬酸或甲酸或乙二酸等、无机酸如盐酸或硫酸或磷酸制成的盐,按其与赋形剂重量比为1:9的比例加入赋形剂,制成胶囊或颗粒剂。实施例6注射液的制备:按实施例1方法先制得化合物(Ⅰ),以及利用有机酸如酒石酸、或柠檬酸或甲酸或乙二酸等、无机酸如盐酸或硫酸或磷酸制成的盐,按常规加注射用水,精滤,灌封灭菌制成注射液。实施例7无菌粉针的制备:按实施例1方法先制得化合物(Ⅰ),以及利用有机酸如酒石酸、或柠檬酸或甲酸或乙二酸等、无机酸如盐酸或硫酸或磷酸制成的盐,将其溶于无菌注射用水中,搅拌使溶,用无菌抽滤漏斗过滤,再无菌精滤,分装于安瓿中,低温冷冻干燥后无菌熔封得粉针剂。上述实施例的作用在于说明本发明的实质性内容,但并不以此限定本发明的保护范围。本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的实质和保护范围。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1