一种菌落挑选通量提高方法与流程
本发明涉及一种基于改进遗传算法的菌落挑选通量提高方法。
背景技术:
菌落挑选仪是近年来生物技术领域中发展较快的一种智能化仪器,其具有菌落自动识别、自动挑选功能,是实现菌落筛选自动化的基础仪器之一。
在这类菌落挑选仪中,探针模块上集成了多根探针,其挑选过程可以概括为:控制模块控制探针模块移动,使其中一根探针正对需要挑选的一个菌落,用该探针进行挑选后再控制探针模块移动、微调,使另一根探针正对需要挑选的下一个菌落并进行挑选,如此往复,对所有需要挑选的菌落进行一一挑选。
但是,上述挑选过程中,探针模块需要移动、微调才能使探针正对需要挑选的菌落,如果需要挑选的菌落数量较多,则探针模块移动及微调的路径也相对较长,使得探针模块的移动耗费较多时间,其挑选通量尚不够理想。
现有技术中,对菌落挑选仪的改进主要集中在机械结构方面,例如,采用更加先进的三维磁力导轨来提高探针模块的移动速度等。这样的改进没有缩短探针模块的实际移动路径,并且使得菌落挑选仪的成本大大提升。而目前尚未有从软件或计算角度对探针模块的控制进行改进,使得探针模块的移动路径最短的方法。
遗传算法是一种常用计算模型,用于对一些复杂问题进行求解。其主要流程是设定一个初始种群,该初始种群包含一定数量的染色体,其中每个染色体对应着问题的一个解。随后模拟自然界生物进化的过程,对初始种群进行遗传迭代操作(包括交叉、变异等),其种群中的遗传信息被不断优化,最后得到的种群中即包含问题的近似最优解。
这种遗传算法具有自适应、自学习的特性,并且能够基于问题全局进行求解,因此在一些复杂问题的解决中能够起到很好的作用。但是,传统的遗传算法在面对实际问题时,其编码操作通常较为困难。并且,遗传算法对初始种群的优劣较为敏感;传统的交叉操作对问题适应性差,使得收敛速度较慢;传统的变异操作容易破坏优秀染色体,或者难以使染色体种群达到足够的变异程度;选择算子设置不当容易使种群陷入局部解。
由于存在上述多种缺陷,即使将遗传算法直接应用于菌落挑选仪的改进,也难以达到使探针模块路径大大缩短、提高挑选通量的目的。
技术实现要素:
为解决上述问题,本发明对传统遗传算法进行了改进,并提出了一种基于改进遗传算法的菌落挑选通量提高方法,采用了如下技术方案:
本发明提供一种菌落挑选通量提高方法,应用于菌落挑选仪中用于使探针模块的挑选路径缩短从而提高挑选通量,其特征在于,包括如下步骤:
步骤1,对需要挑选的菌落及探针模块中的探针进行编号,得到菌落编号以及探针编号,根据探针与菌落的空间位置建立挑选路径的数学模型,其中,需要使用的探针的数量与菌落一致,均为n个;
步骤2,将菌落编号以及探针编号依次排列,得到初始染色体,该初始染色体是长度为2n的实数序列,其中前n个实数为菌落序列,后n个实数为探针序列,菌落编号以及探针编号依次匹配成对;
采用随机插入的方式得到2m个染色体,该2m个染色体的集合为初始种群,其中m为5的倍数,此时迭代次数为0;
步骤3,对父代种群进行迭代操作,得到子代种群,并计算该子代种群中每个染色体的适值,迭代次数加1;
步骤4,用步骤3得到的子代种群替换父代种群,重复步骤3,直到迭代次数达到预定次数,得到最终种群;
步骤5,从最终种群中选择适值最大的染色体作为结果输出,使菌落挑选仪按照结果中菌落及探针的匹配进行菌落挑选,
其中,在步骤3中,迭代操作包括如下子步骤:
步骤3.1,按照下述式(1)计算初始种群中每个染色体的适值,并将初始种群作为父代种群,
式(1)中,i为探针模块移动的次数,Si为探针模块在第i次移动时所移动的距离;
步骤3.2,采用适值分组随机复制策略得到2m个染色体,该2m个染色体的集合为第一子代;
步骤3.3,对步骤4.1得到的第一子代进行交叉操作,得到第二子代;
步骤3.4,变异操作,将步骤4.2中得到的第二子代中的全部染色体按适值排序,求出所有染色体的平均适值fitavg、最大适值fitmax以及最小适值fitmin。遍历所有染色体并进行以下操作,得到子代种群:
在当前染色体的适值高于平均适值的情况下,将预设的固定变异率作为基准值进行随机值判定,若判定成功则进行染色体变异并用变异后的染色体代替当前染色体,若判定失败则跳过;
在当前染色体的适值低于平均适值的情况下,根据下述式(2)计算得到变异率系数
式(2)中,fiti为当前染色体的适值,以计算得到的变异率系数作为基准值进行随机值判定,若判定成功则进行染色体变异并用变异后的染色体代替当前染色体,若判定失败则跳过。
发明作用与效果
本发明提供的菌落挑选通量提高方法中,由于将菌落及探针分别编号、一一对应形成染色体的序列,使得每个染色体都与菌落及探针的一种匹配相对应,使得遗传算法对菌落挑选挑选问题的编码及解码过程简化;本发明还对传统遗传算法进行了改进,使得改进后的遗传算法具有收敛速度快、能规避早熟等优点,因此采用本发明的菌落挑选通量提高方法能够对菌落挑选仪的探针模块控制进行优化,使得挑选过程中探针模块移动路径缩短,从而提高菌落挑选通量。
附图说明
图1是本发明的整体流程图;
图2是本发明实施例的初始染色体序列图;
图3是本发明实施例的染色体序列及适值计算示意图;
图4是本发明实施例的交叉操作示意图;
图5是本发明实施例的变异操作示意图。
具体实施方式
以下结合附图以及实施例来说明本发明的具体实施方式。
<实施例>
在本实施例中,采用探针模块中的8根探针对8个空间位置确定的菌落进行挑选。
图1是本发明的整体流程图,如图1所示,本实施例的菌落挑选通量提高方法包括如下步骤:
步骤1,对8个菌落及8根探针进行编号,得到菌落编号以及探针编号,根据探针与菌落的空间位置建立挑选路径的数学模型。在本实施例中,该8个菌落的编号为8个实数,即1~8;8个探针的编号也为8个实数,即1~8。
其中,挑选路径的数学模型为菌落挑选仪中,控制模块根据探针与菌落的匹配得出的数学模型,即、将探针和菌落的匹配关系输入该数学模型进行计算,即可得出探针模块的移动路径;该移动路径包括探针模块的移动次数以及每次的移动距离。
步骤2,将菌落编号以及探针编号依次排列,得到初始染色体。在本实施例中,菌落编号按数字从小到大排列从而形成菌落序列,同样地,探针编号也按照数字从小到大排列从而形成探针序列。
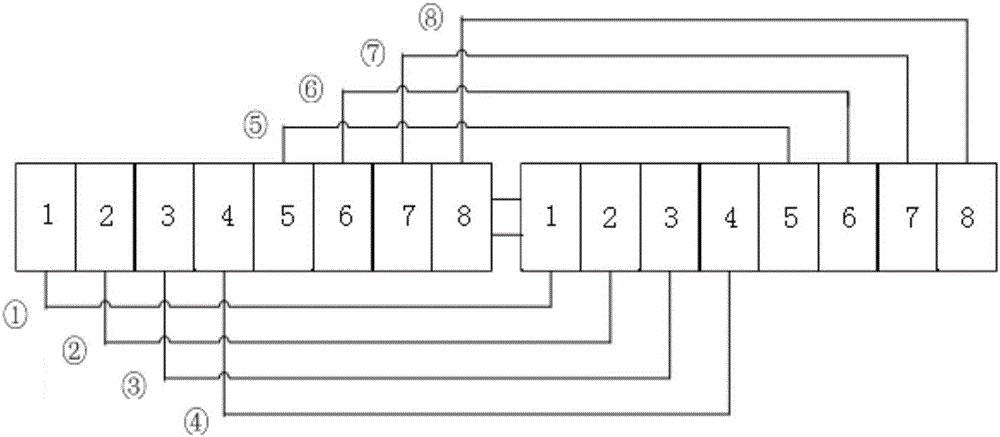
图2是本发明实施例的初始染色体序列图。
如图2所示,初始染色体是长度为16的实数序列,其中前8个实数为菌落序列,每一位数字都是一个菌落编号;后8个实数为探针序列,每一位数字都是一个探针编号。其中,菌落序列中的各个菌落编号与探针序列中的各个探针编号按顺序一一匹配:菌落序列的第一位与探针序列的第一位匹配,形成匹配①;菌落序列的第二位与探针序列的第二位匹配,形成匹配②。以此类推,该染色体中总共具有8对菌落与探针的匹配,分别为匹配①、②、③、④、⑤、⑥、⑦、⑧。
对初始染色体采用随机插入的方式得到2m个染色体,该2m个染色体的集合为初始种群,其中m为5的倍数,设定一个迭代次数并将此时的迭代次数记为0。
上述过程中,随机插入包括如下子步骤:
步骤2.1,在初始染色体中随机选择一个菌落编号及一个探针编号,分别作为插入菌落编号及插入探针编号;
步骤2.2,将菌落序列中,位于插入菌落编号后面的菌落编号依次替换前面的菌落编号并将插入菌落编号插入最后,将探针序列中,位于插入探针编号后面的探针编号依次替换前面的探针编号并将插入探针编号插入最后,如此使得初始染色体原有的序列被打乱,形成与初始染色体不同的一个染色体;
步骤2.3,在重复步骤2.1~2.2预定次数后,对得到的染色体进行记录,在本实施例中,该预定次数为20次;
步骤2.4,重复步骤2.3 100次,得到100个染色体,该100个染色体的集合即为初始种群。
步骤3,将初始种群作为父代种群进行迭代操作,并将迭代次数加1,其中迭代操作包括如下子步骤:
步骤3.1,按照下述式(1)计算初始种群中每个染色体的适值fit:
式(1)中,i为探针模块移动的次数,Si为探针模块在第i次移动时所移动的距离,在本实施例中,n=8。此外,下文中所有的染色体适值均按照此式(1)计算。
图3是本发明实施例的染色体序列及适值计算示意图。
图3所示的为初始种群中的一个染色体。如图3所示,该染色体的长度及匹配数量与初始染色体相同,但由于此前进行的随机插入操作,其中菌落序列、探针序列的排列及菌落编号与探针编号的匹配均与初始染色体不同。
以该染色体为例,其适值的计算方法为:假定挑选开始前,探针模块初始化后位于初始位置,则探针模块移动到匹配①实现的位置为S1,即、当探针模块开始按照菌落挑选仪的控制程序移动,直到使得编号为2的菌落与编号为8的探针在空间上对应,此时编号为8的探针能够对编号为2的菌落进行挑选,探针从初始位置移动到此状态所进行的位移为S1;继续地,从匹配①的状态移动到匹配②实现的状态,探针模块进行的位移为S2。以此类推,探针模块按照该染色体所包含的匹配来使得探针及菌落在空间上一一对应,完成8个探针对8个菌落的挑选,在该挑选过程中,探针模块所移动的总路径为S1+S2+…+S8,即其中n=8,再将该总路径的值代入式(1),即可计算得到该染色体的适值fit。下文中,各匹配的实现均指该匹配中,相应探针编号的探针与相应菌落编号的菌落在空间上对应的实现。
步骤3.2,采用适值分组随机复制策略得到100个染色体,该100个染色体的集合为第一子代,适值分组随机复制策略具体为:
将父代种群中的全部染色体按照适值从高到低排列并分为五组,第一组包含适值排名为前10的染色体,第二组包含适值排名为10~40的染色体,第三组包含适值排名为40~80的染色体,第四组包含适值排名为前80~90的染色体,第五组包含剩余的染色体。由于染色体适值可能有相同的,因此每组中包含的染色体数量可能大于排名位数,例如,第一组包含适值排名为前10的染色体,但其中染色体的数量可能大于10个。
对排在第一位(即适值最高)的染色体进行复制,在第一组中随机选择10个染色体并进行复制,将第一组和第二组合并,从中随机选择40个染色体并进行复制,在第三组中随机选择40个染色体并进行复制,在第四组中随机选择9个染色体并进行复制。经过上述复制,得到的复制染色体数量为100,该100个染色体的集合即为第一子代。
步骤3.3,对步骤3.2得到的第一子代进行交叉操作,具体过程为:
步骤3.3.1,将第一子代中的染色体随机两两配对,形成50对染色体,任意选择一对染色体,将该对染色体记为已访问并在其中任意选择一个作为第一父代,另一个作为第二父代;
步骤3.3.2,从第一父代中的菌落序列中随机选择一个菌落编号作为扩展点A,将该菌落编号所匹配的探针编号作为扩展点B,并对A和B进行标记;
步骤3.3.3,从第一父代的菌落序列中找出与A相邻的菌落编号并将找出的菌落编号及其匹配的探针编号的集合记为α,从第二父代的菌落序列中找出与A相邻的菌落编号并将找出的菌落编号及其匹配的探针编号的集合记为β,并进行如下操作:
当时,查找得出α中未被标记的菌落编号Ax及其匹配的探针编号Bx,将Ax及Bx添加至新子代,然后用Ax及Bx替换A及B,并对新的A及新的B进行标记;
当时,定义x=α∪β,找出x中未被标记的菌落编号及其匹配的探针编号,分别将找出的菌落编号及其匹配的探针编号依次记为Ai以及Bi,其中i=1、2、……t,t≤4,对探针模块从A-B匹配实现的位置开始移动直到Ai-Bi匹配实现的位置时,探针模块所移动的距离Li进行计算,得到L1、L2、……Lt,从L1、L2、……Lt中选出Li值最小的一个并将该最小的Li值记为Lmin,Lmin所对应的Ai及Bi记为Amin及Bmin,将该Amin及Bmin加入新子代并用Amin及Bmin替换A及B形成新的A和新的B,并对该新的A及新的B进行标记;
图4是本发明实施例的交叉操作示意图。
图4a示出了的情况。如图4a所示,此时第一父代中,扩展点A为第5位,其菌落编号为8,扩展点B与扩展点A匹配,在图中为第13位,其探针编号为3。在第一父代中,A相邻的菌落编号为3和6,其对应的匹配为3-7、6-8;在第二父代中,扩展点A在第4位,其相邻的菌落编号为3和4,对应的匹配为3-7、4-8。由此,集合α包含3-7、6-8,集合β包含3-7、4-8。由于α与β有交集,即此时将其交集中的匹配,即3-7添加至新子代中,并用3、7替换原有的扩展点A及扩展点B,对该3、7进行标记。
图4b示出了的情况。如图4b所示,此时第一父代中扩展点A为第5位,菌落编号8,扩展点B为第13位,菌落编号3,A相邻的菌落编号为3和6,其对应的匹配为3-7、6-8;第二父代中,A为第4位,相邻的菌落编号为3和4,对应匹配为3-6、4-8。
此时,集合α包含3-7、6-8,集合β包含3-6、4-8。由于α与β并无交集,因此将α与β合并形成并集χ,该并集χ最多包含四个匹配。在图4b中,χ包含3-7、6-8、3-6、4-8四个匹配。由于这四个匹配中,6-8及4-8已经被标记,因此选择3-7、3-6两个匹配,分别将其中的菌落编号记为A1、A2,其对应的探针编号记为B1、B2。将A和B,即菌落编号8和探针编号3匹配实现的位置作为移动起始点,将A1和B1匹配实现的位置作为移动终止点,根据菌落挑选仪中的控制程序,计算探针模块在从移动起始点移动到移动终止点所移动的距离L1。以此类推,计算出A2和B2所对应的L2。在图4b所举的例子中,L1>L2,因此将L2所对应的菌落编号A2以及探针编号B2,即3-6这一对匹配添加进新子代,并用A2以及B2替换A和B形成新的A和B,再进行接下来的步骤。
步骤3.3.4,重复步骤3.3.3,直到新子代的长度达到16;
步骤3.3.5,将第一父代及第二父代的位置交换,重复步骤4.2.2~步骤3.3.4,得到另一个长度为16的新子代;
步骤3.3.6,从50对染色体中任意选择一对未访问的染色体,从该对染色体中任意选择一个作为第一父代,另一个作为第二父代,重复步骤3.3.2~步骤3.3.5,再次得到两个长度均为16的新子代;
步骤3.3.7,重复步骤3.3.6,直到第一子代中所有染色体对均被记为已访问,如此即完成了所有染色体对的交叉操作,得到数量为100的新子代,该新子代的集合为第二子代;随后进行后续操作。
步骤3.4,变异操作,将步骤3.3中得到的第二子代中的全部染色体按适值排序,求出所有染色体的平均适值fitavg、最大适值fitmax以及最小适值fitmin,遍历所有染色体(即、第二子代中的100个染色体)并进行以下操作,得到子代种群:
在当前染色体的适值高于平均适值的情况下,将预设的固定变异率作为基准值进行随机值判定,若判定成功则进行染色体变异并用变异后的染色体代替当前染色体,若判定失败则跳过,在本实施例中,该固定变异率为20%;
在当前染色体的适值低于平均适值的情况下,根据下述式(2)计算得到变异率系数
式(2)中,fiti为当前染色体的适值。以计算得到的变异率系数作为基准值进行随机值判定,若判定成功则进行染色体变异并用变异后的染色体代替当前染色体,若判定失败则跳过。
上述过程中,随机值判定的规则为:生成一个随机数,当该随机数的值大于基准值时则判定成功,当该随机数的值小于或等于基准值时则判定失败。其中,随机数的取值范围为0~1。
此外,变异操作的策略为:当前染色体的适值大于预设值时进行同步变异,当前染色体的适值小于或等于预设值时进行异步变异,即、根据当前染色体的适值高低,进行不同种类的变异操作。
图5是本发明实施例的变异操作示意图。
图5a示出了同步变异的操作过程。如图5a所示,当前染色体的适值大于预设值,因此进行同步变异:从当前染色体的菌落序列中随机挑选出两个菌落编号3和6,分别记为a1和a2,找出当前染色体探针序列中,分别与a1和a2匹配探针编号4和6,分别记为b1和b2。将a1与a2之间的实数序列反转,同时将b1与b2之间的实数序列反转,即得到变异后的染色体。
图5b示出了异步变异的操作过程。如图5b所示,当前染色体的适值小于预设值,因此进行异步变异:从当前染色体的菌落序列中随机挑选出两个菌落编号8和6,分别记为a1和a2,然后从当前染色体的探针序列中随机挑选出两个探针编号6和5,分别记为b1和b2。将a1与a2之间的实数序列反转,同时将b1与b2之间的实数序列反转,即得到变异后的染色体。
经过上述步骤迭代操作步骤,由原来的父代种群得到了子代种群,该子代种群的数量与父代种群相同,均为100。
步骤4,用步骤3得到的子代种群替换父代种群,重复步骤3,直到迭代次数达到预定次数,得到最终种群。在本实施例中,迭代次数为500次。
步骤5,从最终种群中选择适值最大的染色体作为结果输出,使菌落挑选仪按照结果中菌落及探针的匹配及其顺序进行菌落挑选。
实施例作用与效果
本实施例提供的菌落挑选通量提高方法中,由于将菌落及探针分别编号、一一对应形成染色体的序列,使得每个染色体都与菌落及探针的一种匹配相对应,使得遗传算法对菌落挑选挑选问题的编码及解码过程简化;本实施例还对传统遗传算法进行了改进,使得改进后的遗传算法具有收敛速度快、能规避早熟等优点,因此采用本实施例的菌落挑选通量提高方法能够对菌落挑选仪的探针模块控制进行优化,使得挑选过程中探针模块移动路径缩短,从而提高菌落挑选通量。
此外,本实施例中,迭代操作首先包含适值分组随机复制的过程,使得适值更高的染色体有更多机会被复制至下一代,对下一代进行优化,同时,适值更低的染色体同样有一定机会被复制至下一代,避免陷入局部解;交叉操作中引入了目标点连接信息算子,即、当α与β的交集不为空时,直接将交集中的匹配复制至下一代,使得优秀染色体中共有的片段能够尽可能传至下一代;变异操作中对于较优染色体采用一个较低的固定变异率,使得优秀染色体尽量减少变异,并且,适值高于预定值的采用同步变异、适值低于预定值的采用异步变异,也使得优秀的染色体中的匹配尽量保留。如此的迭代操作克服了传统遗传算法中的多种缺点,使得本实施例的方法能够达到更好的效果。
上述实施例仅用于说明本发明的具体实施方式,本发明的方法的范围不限于上述实施例的范围。
例如,在上述实施例中,初始染色体的生成采用菌落编号及探针编号依次排序的方式来组成菌落序列和探针序列,但在本发明中,菌落编号也可随机排列来形成菌落序列,探针编号也可随机排列来形成探针序列。此外,在上述实施例中,初始染色体在进行20次随机插入后形成的染色体被记录成为初始种群中的一个,但在本发明中,该随机插入的次数也可以是其他次数;上述实施例中,初始种群的数量为100,迭代次数为500次,但在本发明中,该初始种群的数量也可以是其他数量,迭代次数也可以是其他次数。
- 还没有人留言评论。精彩留言会获得点赞!