一种识别猕猴桃杂交亲本对子代基因组贡献比例的方法与流程
本发明属于植物遗传资源评价和生物技术领域,具体涉及一种识别猕猴桃杂交亲本对子代基因组贡献比例的方法和应用,即用于猕猴桃杂交组合中亲本对杂交后代基因组贡献比例的分析,从而形成对杂交子代性状变异幅度和方向的预测,提升杂交子代的早期筛选效率,减少土地和人力成本,促进杂交新品种的快速培育。
背景技术:
作物杂交育种是种质资源改良和新品种培育的重要手段,通过杂交通常可以整合双亲优异性状并进一步产生新的跨亲特征,在产量、生态适应性和抗性等方面形成广泛的变异。猕猴桃(Actinidia chinensis Planch.)是原产我国的重要新兴水果,因富含维生素C等多种营养成分而受到消费者的广泛喜爱。近年来,猕猴桃人工杂交育种取得了较大进展,一些杂交新品种的出现,无论是在品质特性和种植适应性等方面都优于其他常规品种,促进了猕猴桃产业发展和商品销售。同时,猕猴桃本身为雌雄异株多年生植物,种间自然杂交也十分频繁,形成了大量自然杂交种质资源可以用来进行遗传评价和育种应用。在猕猴桃的杂交资源评价过程中,确定杂交亲本对子代的基因组贡献比例,可以有效的预测其杂交后代性状变异的幅度和方向,从而形成早期杂交子代的人工筛选,大大提升资源发掘利用的效率。然而,迄今为止,缺乏准确有效的技术方法来进行杂交基因组比例的预测,使得猕猴桃杂交育种过程中杂交资源的早期选择存在极大的盲目性,并浪费了大量的人力物力成本,不利于杂交新品种的快速培育和创新。
技术实现要素:
本发明的目的是针对猕猴桃杂交资源早期选择效率低下的问题,提供一种识别猕猴桃杂交亲本对子代基因组贡献比例的方法,其能用于猕猴桃杂交后代性状变异幅度和方向的预测,促进杂交资源早期选择效率。
本发明通过整基因组范围单碱基变异(SNPs)标记的获取以及一定大小基因组窗口的划分,按基因组窗口构建猕猴桃杂交双亲和杂交子代及一个远缘猕猴桃外类群的四类群(four-taxon)最大可能(maximum likelihood)亲缘关系树(定义为基因树),然后在整基因组范围内统计支持杂交后代与其中一个亲本聚类的基因树数量,根据各自亲本与杂交子代聚类的基因树数量所占的比例形成杂交亲本对杂交子代基因组贡献比例的分析预测。
本发明包括待分析杂交子代与杂交双亲的基因组低深度测序,将测序数据比对到猕猴桃参考基因组获取SNPs变异,基于变异信息的10-kb非重叠基因组窗口划分,然后对每个窗口内所有变异位点,估计其对杂交亲本与子代亲缘关系支持的对数可能性(site-wise log likelihoods),基于位点可能性估计值构建该窗口的最大可能性进化发育关系,即该窗口的基因树,最后利用整基因组范围内基因树对杂交双亲的支持率预测其对杂交子代基因组内容的贡献比例。
本发明的识别猕猴桃杂交亲本对子代基因组贡献比例的方法,其特征在于,包括以下步骤:
A、对猕猴桃杂交双亲样本和子代样本及远缘猕猴桃外类群样本进行基因组低深度测序,获得各样本的基因组;
B、测序数据的参考基因组比对和单碱基变异信息获取:将各样本的基因组比对到中华猕猴桃参考基因组,对基因组比对序列进行基因型构建和变异信息发掘,同时对所获得的单碱基变异进行过滤,去掉不可靠的变异信息位点;
C、基于单碱基变异的基因组窗口划分和对数可能性估计:基于步骤B获得的变异信息,对各样本基因组进行非重叠的窗口划分,所有4个样本间的窗口划分起始点保持一致,对所划分的窗口进行进一步的过滤,包括去掉参考基因组上拥有连续N的窗口,去掉变异质量值低于20的窗口,去掉具有大量插入缺失变异位点的窗口;
对所有窗口的变异位点对数可能性进行计算;
D、分窗口最大可能基因树构建和置信区间估计:将所有获得的变异位点对数可能性值输入到Consel工具包的makermt模块进行标准化分析生成基因树,得到的结果再进一步输入到该工具包的consel模块进行大约无偏AU检测,通过该检测的P值判断窗口最大可能支持的样本亲缘关系(基因树)种类,对于所有窗口形成的基因树进行95%的置信区间估计;
E、杂交双亲在基因树水平与子代遗传关系统计及基因组贡献比例预测:统计整个基因组范围内全部窗口的样本亲缘关系的支持比例及其置信区间,据此判断猕猴桃杂交亲本对子代基因组的贡献比例。
所述的步骤C的基于步骤B获得的变异信息,对各样本基因组进行非重叠的窗口划分,优选是基于步骤B获得的变异信息,对各样本基因组进行10-kb非重叠的窗口划分。
相比现有技术,本发明有益效果如下:
①本发明利用整基因组变异信息解析杂交亲本与杂交子代的进化遗传关系,使得对杂交子代特征性状的预测具有较高的准确性;
②使用该方法可以在杂交子代形成的童期对其可能存在的表型特征变异的幅度和方向进行预测,可以极大的促进杂交株系的早期筛选,节省人力物力成本,并大大提高资源发掘利用效率;
③本方法独立于检测的植物类群,可应用到其他植物或者农作物的杂交基因组遗传比例的估计,特别是可推广到不易在短时间内观测到杂交子代重要农艺性状特征的多年生作物。
附图说明:
图1是本发明的流程图;
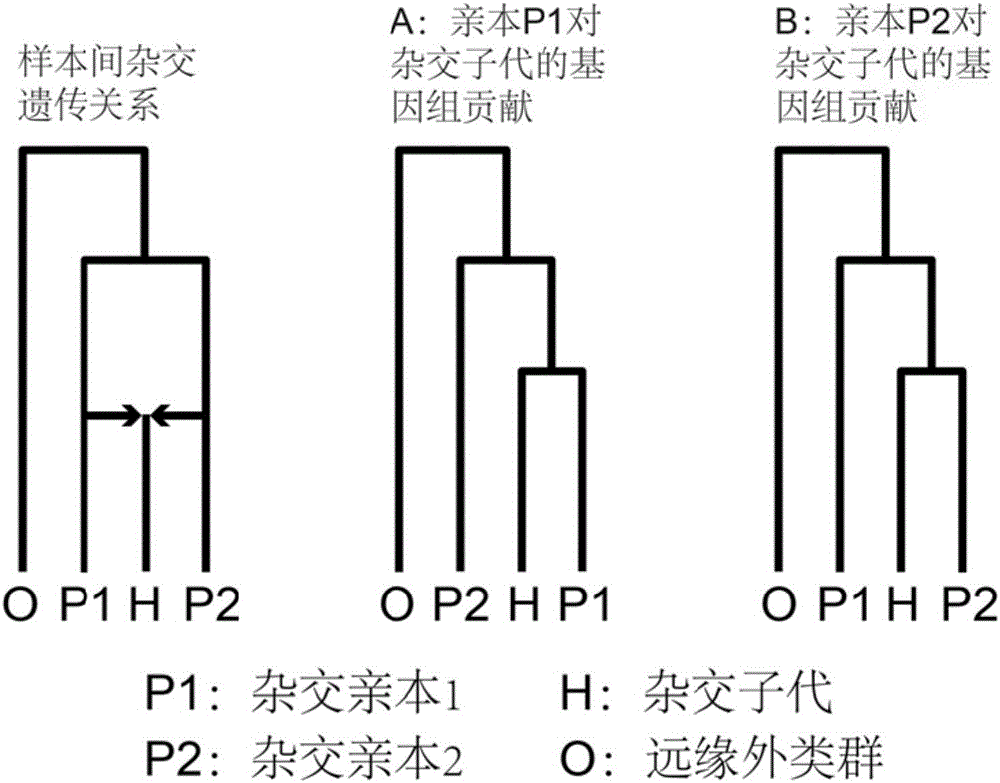
图2是本发明所采用的四类群亲缘关系树。
具体实施方式:
以下实施例是对本发明的进一步说明,而不是对本发明的限制。
实施例1:本实施例的识别猕猴桃杂交亲本对子代基因组贡献比例的方法,其步骤如下:
1.如图1步骤①对杂交双亲和子代及远缘外类群进行基因组低深度测序。
选择待分析的猕猴桃杂交双亲和子代样本及一个亲缘关系上与该杂交组合的所有材料相对远缘的猕猴桃外类群样本,提取每个样本的基因组总DNA约5μg左右,然后构建采用Illumina测序平台提供的试剂盒构建180bp左右的小片段文库并在其Hiseq2000平台测序。总共测序数据量达到3.5Gb左右,即得各样本的整体测序深度为5倍的猕猴桃基因组。
对测序片段使用FastQ软件进行序列过滤。过滤标准采用软件自带的缺省数值。
2.如图1步骤②测序数据的参考基因组比对和单碱基变异信息获取。利用免费的Stampy软件将各样本的猕猴桃基因组测序数据比对到现有的中华猕猴桃参考基因组,比对参数为缺省值设置。利用免费的基因组分析工具GATK对基因组比对序列进行基因型构建和变异信息发掘。同时参照宽泛研究所(Broad Institutions)的建议过滤标准对所获得的单碱基变异进行严格过滤,去掉不可靠的变异信息位点,包括去掉QUAL值小于40或者DP值大于3300的变异位点,去掉低频突变位点(<0.01)以及5bp范围内的连续变异位点。
3.如图1步骤③基于单碱基变异的基因组窗口划分和对数可能性估计。基于步骤2获得的变异信息,对猕猴桃样本的基因组(猕猴桃杂交双亲和子代样本及一个亲缘关系上与该杂交组合的所有材料相对远缘的猕猴桃外类群样本)进行10-kb非重叠的窗口划分,所有四个样本间的窗口划分起始点保持一致。同时,对所划分的窗口进行进一步的过滤,包括去掉参考基因组上拥有连续N的窗口,去掉变异质量值低于20的窗口,去掉具有大量插入缺失变异位点的窗口。
然后利用免费的RAxML软件对所有窗口的变异位点对数可能性进行计算,计算时采用General Time Reversible模式和核酸替代率的gamma分布模式,同时使用-fg选项。
4.如图1步骤④分窗口最大可能基因树构建和置信区间估计。将所有获得的变异位点对数可能性值输入到免费的Consel工具包的makermt模块进行标准化分析生成基因树(基因树有3种,具体如图2所示),得到的结果再进一步输入到该工具包的consel模块进行大约无偏AU检测。该检测的P值反映了该基因组的窗口最大可能支持哪一种样本间的亲缘关系(图2的A或者B)。本实施例采用P>0.95作为该窗口最大可能基因树的选择标准。
对于所有窗口形成的基因树,利用免费的R软件(www.r-project.org)进行95%的置信区间估计,使用1000次非参数重取样。
5.如图1步骤⑤杂交双亲在基因树水平与子代遗传关系统计及基因组贡献比例预测。统计整个基因组范围内全部窗口如图2所示的A或者B两种样本亲缘关系(基因树)的支持比例及其置信区间。在0–100%的理论支撑比例区间,本实施例设置双亲的支持比例处于40–60%区间的为大概同等的杂交子代基因组贡献率,例如大多F1代杂合子类型;而处于其他区间的则为杂交子代的基因组遗传内容更偏向于遗传某一个亲本,反映为两者之间更为相近的进化遗传关系,这种情况多相关于杂交过程中的不对称基因流或者经历了回交过程的杂交后代。
二、应用
按照步骤一的方法,本实施例共分析了猕猴桃人工杂交组合8个,涉及的杂交双亲猕猴桃物种包括柱果猕猴桃、毛花猕猴桃、中华猕猴桃、硬齿猕猴桃、山梨猕猴桃和软枣猕猴桃等,其双亲对杂交子代的基因组贡献比例在4.14–95.71%之间(如下表1所示),其中4对杂交组合的支持率处于40–60%区间,反映了大概同等的双亲对杂交子代的基因组贡献,而其他的处于这个区间之外,则多相关于渐渗型基因流或者回交,反映了一个亲本对杂交后代基因组的优势贡献。这些结果与我们的表型观察值保持高度的一致,同时也相关于杂交过程中存在的回交行为。
表1实施例检测的杂交亲本对杂交后代基因组贡献的比例
由此可见,本发明能准确的反映杂交过程中猕猴桃杂交亲本对杂交后代基因组遗传物质的贡献比例。由于该比例与所观测的表型值和育种过程具有高度一致性,因而可以用该比例来预测猕猴桃杂交后代变异的幅度和方向,促进杂交后代早期筛选,提高猕猴桃杂交育种的效率。
- 还没有人留言评论。精彩留言会获得点赞!