一种多拉司琼异构体或其盐的制备方法与流程
本发明属于药物合成技术领域,更具体地讲,涉及多拉司琼异构体杂质或其盐的制备方法。
背景技术:
多拉司琼(Dolasetron mesylate)是一种高度选择性5-羟色胺(5-HT3)受体拮抗剂,由德国Hoechst Marion Roussel公司研发成功,本品口服和静脉注射对防治癌症化疗引起的恶心或呕吐均有效。5-HT3受体拮抗剂联用地塞米松或其他皮质激素对预防癌症化疗引起的急性呕吐是最有效的复合治疗方案。美国FDA建议,本品也可用于预防和治疗术后恶心或呕吐。
目前,关于多拉司琼合成工艺的文献报道较少,欧洲专利EP0329902对多拉司琼的合成方法进行了阐述。根据该专利公布的合成路线,式17的化合物经硼氢化钠还原制备式18的化合物,这步反应受桥环以及船式构象的影响,还原后羟基优势构象处于直立键;但受反应条件的影响,在还原过程中可能产生羟基处于平伏键的异构体,从而导致后续式Ⅱ的中间异构体杂质的产生,如下式所示:
式Ⅱ的化合物和其异构体杂质都可以与吲哚-3-甲酸反应,因而根据该路线制备多拉司琼时,会导致多拉司琼异构体杂质(式Ⅰ化合物)的产生,从而影响产品质量,存在较大的安全隐患。
关于多拉司琼的异构体杂质的研究文献很少。专利CN102180878首次公开了式I的多拉司琼异构体杂质,并对其异构体杂质的制备方法进行了详细的描述。该专利的制备方法中,关键步骤在于式(1)的中间体的制备,为得到式(1)的中间体,该专利提供了两种路线,第一条路线需要3步,但是其氢化还原的步骤容易生成与多拉司琼相同构型的杂质,针对该问题,第二条路线设计将生成的与多拉司琼相同构型的杂质用Mitsunobu反应实现直立键羟基的构型翻转,共经过5步反应得到式(1)的中间体。这两种方法都存在反应步骤长和纯度低的不足。
鉴于式I的多拉司琼异构体杂质用现有合成方法具有一定的不足,而其在原料药以及制剂产品检测中又极具重要性,本发明旨在对此杂质的制备方法进行研究,从而为多拉司琼的工艺研究指明方向并保证产品质量和用药安全。
技术实现要素:
为了解决现有技术中存在的问题,本发明的目的是提供一种式I的多拉司琼异构体杂质或其盐的制备方法。本发明的方法所使用的原料易得,合成路线短,产品纯度高。
本发明的一种式I的多拉司琼异构体杂质或其盐的制备方法包括以下步骤:
1)式II的化合物或其盐与RSO2X或CF3COY进行酯化反应,得到式III的中间体;
2)式III的中间体再与式IV的化合物进行取代反应,得到式I的多拉司琼异构体杂质或其盐;
其中,式II的化合物的盐和式I的化合物的盐分别独立的选自氢卤酸盐、硝酸盐、硫酸盐、四氟硼酸盐、草酸盐、枸橼酸盐、酒石酸盐、扁桃酸盐、醋酸盐、甲磺酸盐、对甲苯磺酸盐或樟脑磺酸盐;所述氢卤酸盐包括氢氟酸盐、盐酸盐、氢溴酸盐、氢碘酸盐;
R1=RSO2或CF3CO;
X选自F、Cl、Br、I或RSO3-;
Y选自F、Cl、Br、I或CF3CO2-;
R选自取代或未取代的C1-C6烷基,取代或未取代的C6-C10芳基,其中,所述取代是指被至少一个选自以下组中的基团取代:卤素,被卤素取代或未取代的C1-C3烷基,硝基,氰基。所述烷基包括支链和直链烷基,所述卤素选自F、Cl、Br和I;
M1和M分别独立的选自H或碱金属,所述碱金属包括锂、钠、钾、铷、铯,
本领域的技术人员根据常规知识或本发明的实施例可知,当步骤2)的反应结束后,用酸调节反应液的pH至酸性,或者在反应液为酸性的条件下不作调至碱性处理,即可得到式I多拉司琼异构体杂质的对应盐。
根据本发明的一个实施例,在步骤2)反应结束后,反应液本身为酸性,不调节pH即可得到式I对应的盐。
本发明的方法选用式II的化合物为原料,该化合物构型与多拉司琼相同,是制备多拉司琼所需的原料,因此市场很容易获得,而不需要根据现有技术的方法,例如专利CN102180878的方法,经过多个步骤先制备式II的异构体后再酯化。根据本发明的方法可以将式II的化合物在两步成酯的过程中完成构型翻转制得高纯度的式I的多拉司琼异构体杂质。
作为优选,式II和式I的化合物的盐分别独立的选自盐酸盐、氢溴酸盐、硝酸盐、硫酸盐、四氟硼酸盐、甲磺酸盐和对甲苯磺酸盐;更优选为盐酸盐、氢溴酸盐、硝酸盐、硫酸盐、四氟硼酸盐和酒石酸盐。
作为优选,X选自Cl和RSO3-。
作为优选,Y选自Cl和CF3CO2-。
作为优选,R选自取代或未取代的C1-C3烷基,取代或未取代的苯基,其中所述取代是指被至少一个选自以下组中的基团取代:卤素,硝基,甲基和乙基。所述烷基包括支链和直链烷基,所述卤素选自Cl和Br;作为更优的选择,R选自甲基、乙基、三氟甲基、苯基、对甲苯基、对氯苯基、对溴苯基、对硝基苯基。
M1和M分别独立的选自H、锂、钠、钾和铯。
本领域的技术人员可知,步骤1)和2)的反应在碱性条件下更利于反应的进行,所述碱性条件采用的碱包括无机碱和有机碱。
所述无机碱包括但不限于氢化钠、氢化钾、氨基钠、氢氧化钾、氢氧化钠、氢氧化锂、碳酸钾、碳酸钠、碳酸铯、碳酸氢钠、碳酸氢钾中的一种或多种,所述有机碱包括但不限于三乙胺、二异丙基乙胺、吡啶、N-甲基吗啉、4-二甲氨基吡啶、二氮杂二环(DBU)、甲醇钠、乙醇钠、叔丁醇钾、叔丁醇钠中的一种或多种。作为更优的选择,所述无机碱选自碳酸钾,所述有机碱选自三乙胺、二异丙基乙胺和叔丁醇钾。
步骤1)和步骤2)的反应可以在选自酮类溶剂、烷烃类溶剂、芳烃类溶剂、醚类溶剂、腈类溶剂、酰胺类溶剂和亚砜类溶剂中的一种或多种溶剂中进行,所选溶剂对反应物具有较好的溶解性,并且不影响反应即可,这些溶剂包括但不限于丙酮、四氢呋喃、吡啶、乙腈、二氯甲烷、二甲基乙酰胺,二甲基甲酰胺(DMF)、乙醚、N-甲基吡咯烷酮、DMSO、甲苯、乙苯等。
在本发明的一些实施例中,步骤1)所用的溶剂选自二氯甲烷、DMSO、二甲基甲酰胺(DMF)中的一种或多种,步骤2)所用溶剂选自四氢呋喃、DMF种的一种或两种。
作为优选,式IV的化合物为碱式盐时,可以通过吲哚-3-甲酸与碱原位生成制得,所述碱选自氢化钠、氢化钾、氨基钠、氢氧化钾、氢氧化钠、氢氧化锂、碳酸钾、碳酸钠、碳酸铯、碳酸氢钠和碳酸氢钾。
除非相互矛盾或者相互排斥,本发明上述的优选方式均可以单独采用,或者组合采用。
本发明通过一种新的合成路线定向合成式I的多拉司琼异构体杂质,用于检测甲磺酸多拉司琼原料药或其制剂的质量,所用合成方法具有工艺原料易得、合成路线短、产品纯度高、反应条件温和等优点,能够为甲磺酸多拉司琼的工业化生产监控以及质量研究提供保障。
附图说明
图1为实施例1制得的多拉司琼异构体杂质(式Ⅰ化合物)的质谱图;
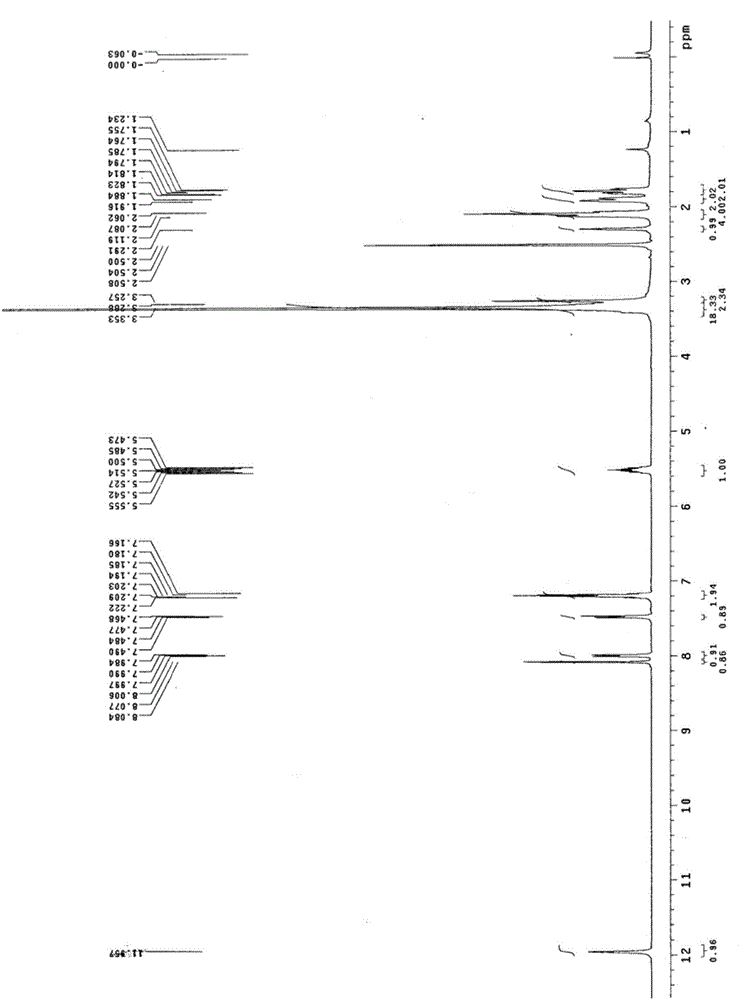
图2为实施例1制得的多拉司琼异构体杂质(式Ⅰ化合物)的核磁共振氢谱图;
图3为实施例1制得的多拉司琼异构体杂质(式Ⅰ化合物)与多拉司琼的HPLC分离图谱。
具体实施方式
本说明书中公开的所有特征,或公开的所有方法或过程中的步骤,除了互相排斥的特征和/或步骤以外,均可以以任何方式组合。
本说明书中公开的任一特征,除非特别叙述,均可被其他等效或具有类似目的的替代特征加以替换。即,除非特别叙述,每个特征只是一系列等效或类似特征中的一个例子而已。
下面对本发明多拉司琼异构体杂质的制备方法进行详细说明。
实施例1:式Ⅰ化合物制备
依次将20mL干燥二氯甲烷、2.0g式Ⅱ化合物、2.3mL三乙胺加入单口烧瓶中,冰浴条件下搅拌降温至0℃,将1mL甲磺酰氯缓慢滴入到反应液中,缓慢升温至室温反应1h;TLC检测反应完全。向反应液中加入20mL水淬灭反应,分液,有机相干燥,浓缩得式Ⅲ化合物(R1=甲磺酰基Ms-)。
将上述式Ⅲ化合物(R1=甲磺酰基Ms-)溶于20mL干燥THF中,搅拌条件下将2.6g式Ⅳ化合物(M1=H,M=K)分批加入反应液中,缓慢升温至80℃反应。TLC监控反应。向反应液中依次加30mL水,二氯甲烷萃取3次,有机层合并,用饱和碳酸钾溶液洗、饱和氯化钠洗涤后,无水硫酸钠干燥、过滤、浓缩得白色固体,柱层析得式Ⅰ化合物为类白色固体约2.1g,HPLC纯度:99.3%。
图1示出了实施例1制备得到的式Ⅰ化合物的质谱图,图2示出了实施例1制备得到的式Ⅰ化合物的核磁共振氢谱图,图3示出了按照CN102183589描述的以硅胶为填充剂、正己烷-异丙醇-二乙胺(30:70:0.1)为流动相,检测波长为285nm的HPLC方法对本实施例的杂质和多拉司琼进行检测的分离图谱。
1H-NMR(d6-DMSO,400MHz):δ=11.95(s,1H),8.08(d,J=2.8Hz,1H),8.01-7.98(m, 1H),7.49-7.47(m,1H),7.22-7.16(m,2H),5.56-5.47(m,1H),3.35-3.26(m,4H),2.29(s,1H),2.12-2.06(m,4H),1.92-1.76(m,4H)ppm。MS-ESI(m/z):325.1550(M+H+)。
实施例2:式Ⅰ化合物制备
依次将20mL干燥N,N-二甲基甲酰胺、2.3g式Ⅱ化合物和2.5g二异丙基乙胺加入单口烧瓶中,搅拌条件下,将2.9g对甲苯磺酰氯分批加入反应液中,室温搅拌反应3h;TLC监测反应完全。向反应液中加入30mL水淬灭反应,乙酸乙酯萃取,分液、有机层再用饱和氯化钠溶液洗涤,无水硫酸钠干燥、过滤、浓缩得式Ⅲ化合物(R1=对甲苯磺酰基Tos-)。
将上述式Ⅲ化合物(R1=对甲苯磺酰基Tos-)溶于20mL干燥N,N-二甲基甲酰胺(DMF)中,搅拌条件下将2.6g式Ⅳ化合物(M1=H,M=Na)分批加入反应液中,缓慢升温至80℃反应。TLC监控反应。向反应液中依次加30mL水,二氯甲烷萃取3次,有机层合并,用饱和碳酸钾溶液洗、饱和氯化钠洗涤后,无水硫酸钠干燥、过滤、浓缩得白色固体,柱层析得式Ⅰ化合物为类白色固体约1.7g,HPLC纯度:98.9%。
实施例3:式Ⅰ化合物制备
依次将20mL干燥吡啶、2.0g式Ⅱ化合物加入单口烧瓶中,冰浴搅拌降温至0℃条件下,将2.8g三氟醋酸酐缓慢滴入反应液中,滴完后,缓慢升温至室温搅拌反应3h;TLC监测反应完全。向反应液中加入30mL水淬灭反应,乙酸乙酯萃取,分液、有机层再用饱和氯化钠溶液洗涤,无水硫酸钠干燥、过滤、浓缩得式Ⅲ化合物(R1=三氟乙酰基)。
将2.1g吲哚-3-甲酸溶于20mL干燥N,N-二甲基甲酰胺(DMF)中,搅拌条件下将2.5g叔丁醇钾分批加入反应液中,搅拌反应30min。将溶有上述式Ⅲ化合物(R1=三氟乙酰基)的DMF溶液加入反应液中,缓慢升温至80℃反应。TLC监控反应。向反应液中依次加50mL水,二氯甲烷萃取3次,有机层合并,用饱和碳酸钾溶液洗、饱和氯化钠洗涤后,无水硫酸钠干燥、过滤、浓缩得白色固体,柱层析得式Ⅰ化合物为类白色固体约1.2g,HPLC纯度:98.6%。
实施例4:式Ⅰ化合物甲磺酸盐制备
依次将20mL干燥二氯甲烷、2.0g式Ⅱ化合物、2.3mL三乙胺加入单口烧瓶中,冰浴条件下搅拌降温至0℃,将1mL甲磺酰氯缓慢滴入到反应液中,缓慢升温至室温反应1h;TLC检测反应完全。向反应液中加入20mL水淬灭反应,分液,有机相干燥,浓缩得式Ⅲ化合物(R1=甲磺酰基Ms-)。
将上述式Ⅲ化合物(R1=甲磺酰基Ms-)溶于10mL干燥N,N-二甲基甲酰胺(DMF)中,搅拌条件下将3.0g式Ⅳ化合物(M1=M=H)加入反应液中,缓慢升温至100℃反应。TLC监控反应。反应液降温至室温后,向反应液中加入30mL乙酸乙酯,搅拌析晶、过滤得类白色固体,结晶纯化得类白色固体约1.1g,HPLC纯度:99.1%。
综上所述,本发明的制备方法具有工艺原料易得、合成路线短、产品纯度高、反应条件温和等优点,能够为多拉司琼的工业化生产监控以及质量研究提供保障。
本发明并不局限于前述的具体实施方式。本发明扩展到任何在本说明书中披露的特征或任何特征的组合,以及披露的任一方法或过程的步骤或其组合。
- 还没有人留言评论。精彩留言会获得点赞!