石蒜科植物忽地笑细胞色素P450还原酶1及其编码基因与应用的制作方法
本发明涉及生物技术和植物生物学领域;更具体地,本发明涉及一种来源于石蒜科植物的细胞色素P450还原酶及其编码基因与应用。
背景技术:
植物细胞色素P450酶(Cytochrome P450enzymes,CYPs)参与大量植物天然产物的生物合成,是天然产物生物合成的关键酶之一,属于亚铁血红素单加氧酶超大家族,催化多种类型的氧化还原反应,具有结构选择性、区域选择性和立体选择性。CYPs的催化活性严格依赖于细胞色素P450还原酶。细胞色素P450还原酶(EC1.6.2.4Cytochrome P450Reductase,CPR)是CYPs催化体系的重要组成部分,属于核黄素蛋白家族,具有黄素腺嘌呤二核苷酸(FAD)结合结构域和黄素单核苷酸(FMN)结合结构域。作为CYPs的氧化还原分子伴侣,细胞色素P450还原酶可以从电子供体(还原力)中获得电子,并通过辅因子FAD和FMN,将获得的电子传递给CYPs,协助CYPs所催化的一系列氧化还原反应。如果没有CPR的存在,CYPs对其底物就没有催化活性。因此,CPR是CYPs催化进行结构专一性、区域专一性和立体专一性生物反应所必需的:CPR催化电子从电子供体(还原力)通过FAD和FMN传递到CYPs的亚铁血红素辅基,协助CYPs催化底物的生物转化。
植物细胞中CYPs的种类数量一般有200多种,而CPR蛋白却只有2-5种。也就是说,植物CPR蛋白在协助植物CYPs进行底物的转化(产物的合成)过程中具有较好的适用性。因此,在研究细胞色素P450酶的催化活性以及利用细胞色素P450酶进行生物转化和生物合成时,如果没有对应物种的CPR,可以使用来源于其他物种的CPR代替。也就是说,CPR在植物天然产物的体内合成、生物转化以及发酵过程中具有重要的作用。
忽地笑(Lycoris aurea)是多年生草本球根药用植物,属于石蒜科石蒜属,富含加兰他敏、石蒜碱、文殊兰碱等石蒜科植物所特有的生物碱(石蒜科生物碱)与其它生物碱、以及其它类型的植物天然产物。这些天然产物具有重要的生物活性和应用价值。例如,加兰他敏作为一种特定的、竞争性的、可逆的乙酰胆碱酯酶抑制剂,在临床上用于治疗阿尔茨海默病(老年痴呆症)。又例如,作为一种苯酚衍生物,对-香豆酸是植物苯丙素类天然产物,具有抗氧化、抗病毒、抗癌及抗炎等多种生物医药活性,用于临床及医药、食品和化妆品等行业。作为一个多功能的药效基团和平台化合物,对-香豆酸还可以衍生出大量具有医药活性的化合物(包括上述在临床治疗轻、中度老年痴呆症的药物-加兰他敏等),具有广泛的应用和需求。在这些天然产物的生物合成过程中,细胞色素P450酶(CYPs)参与催化一步或多步反应,而其催化功能依赖于细胞色素P450还原酶(CPR)。但是,石蒜属植物细胞色素P450还原酶及其编码基因尚未被分离与克隆。因此,本领域有必要开发石蒜属植物忽地笑的细胞色素P450还原酶。该蛋白及其编码基因可通过植物转基因和异源表达的方式协助细胞色素P450酶进行天然产物的生物转化与生物合成。
技术实现要素:
本发明的目的在于提供一种石蒜科植物的细胞色素P450还原酶,所述的细胞色素P450还原酶选自:
(a)氨基酸序列如SEQ ID NO:1所示的蛋白质;或
(b)将SEQ ID NO:1氨基酸序列经过一个或多个(如1-90个)氨基酸残基的取代、缺失或添加而形成的,且具有细胞色素P450还原酶活性的由(a)衍生的蛋白质;或
(c)与SEQ ID NO:1氨基酸序列有至少86%序列相同性,且具有细胞色素P450还原酶活性的由(a)衍生的蛋白质。
本发明SEQ ID NO:1所示的蛋白质是从忽地笑(Lycoris aurea)中分离得到的一种新的细胞色素P450还原酶。为便于表述,将SEQ ID NO:1所示的蛋白质命名为细胞色素P450还原酶LaCPR1。
在一个优选例中,所述的细胞色素P450还原酶活性是指利用还原型烟酰胺腺嘌呤二核苷酸磷酸(NADPH)为电子供体(还原力),传递电子给细胞色素P450酶的替代物(细胞色素c)。
在另一个优选例中,所述的细胞色素P450还原酶活性是指传递电子给细胞色素P450酶(Cytochrome P450enzyme,CYPs),并协助CYPs催化其底物合成产物;例如植物天然产物的生物转化与生物合成。
在另一个优选例中,所述的序列(c)还包括:由(a)或(b)添加了标签序列、信号序列或分泌信号序列后所形成的融合蛋白。
本发明的另一目的在于提供分离的多核苷酸,该多核苷酸是编码所述细胞色素P450还原酶的多核苷酸。
在一个优选例中,该多核苷酸的核苷酸序列如SEQ ID NO:2所示。
应理解,考虑到密码子的简并性以及不同物种密码子的偏好性,本领域技术人员可以根据需要使用合适特定物种表达的密码子。因而,本发明细胞色素P450还原酶的多核苷酸还包括由SEQ ID NO:2所示核苷酸序列经取代、缺失和/或增加一个或几个核苷酸,得到的编码具有细胞色素P450还原酶活性的核苷酸序列。
本发明的又一目的是提供一种载体,它含有所述的多核苷酸。所述的载体是将本发明的编码所述细胞色素P450还原酶的多核苷酸与表达载体可操作地连接,得到能够表达本发明所述细胞色素P450还原酶的重组表达载体或抑制本发明所述细胞色素P450还原酶编码多核苷酸表达的基因沉默载体。
在一个优选例中,该载体是含有编码所述细胞色素P450还原酶的SEQ ID NO:2所示序列的重组表达载体pET28a-LaCPR1。
在另一个优选例中,该载体是含有编码所述细胞色素P450还原酶的SEQ ID NO:2所示序列中第175-2088位多核苷酸的重组表达载体pET28a-LaCPR1(ΔN58)。
本发明的又一目的是提供一种宿主细胞,它含有所述的载体或基因组中整合有所述的多核苷酸。所述的宿主细胞是原核细胞或真核细胞。常用的原核宿主细胞包括大肠杆菌、枯草杆菌、运动假单胞菌和乳酸菌等;常用的真核宿主细胞包括真菌细胞、植物细胞、昆虫细胞和哺乳动物细胞等。所述的真菌细胞包括酵母细胞。将所述的重组表达载体或者基因沉默载体导入所述的适当宿主细胞中,获得表达本发明所述细胞色素P450还原酶的基因工程菌株、转基因细胞系、转基因愈伤、转基因组织、转基因植株或者基因工程植株。
本发明的又一目的是提供所述的细胞色素P450还原酶的用途,用于传递电子给细胞色素P450酶并协助细胞色素P450酶对其底物进行氧化修饰合成产物。
在一个优选例中,所述的细胞色素P450酶是CYP73A;或所述的底物是反式-肉桂酸。
本发明的又一目的是提供一种表达构建物。所述表达构建物包括以下酶的编码基因和/或基因表达盒:
所述的细胞色素P450还原酶;和
细胞色素P450酶;和/或
细胞色素P450酶底物的生物合成酶。
所述基因表达盒是酶在宿主细胞中表达与调控所需要的生物学元件,包括启动子、增强子、衰减子、核糖体结合位点、Kozak序列、内含子和/或转录终止子等;此外,还可包括标签编码序列和/或信号(肽)编码序列等。
在一个优选例中,所述的表达构建物还包括:反式-肉桂酸-4-羟化酶(CYP73A)的编码基因。
在另一个优选例中,所述的表达构建物还包括:反式-肉桂酸-4-羟化酶(CYP73A)的编码基因和苯丙氨酸氨基裂解酶(PAL)编码基因。
在另一个优选例中,当转化大肠杆菌细胞时,所述的表达构建物中,还包含大肠杆菌启动子、大肠杆菌核糖体结合位点和/或大肠杆菌转录终止子等基因表达盒。
本发明的又一目的是提供一种宿主细胞。所述的宿主细胞中包括所述的表达构建物。所述的宿主细胞是原核细胞或真核细胞。常用的原核宿主细胞包括大肠杆菌、枯草杆菌、运动假单胞菌和乳酸菌等;常用的真核宿主细胞包括真菌细胞、植物细胞、昆虫细胞和哺乳动物细胞等。较佳地,所述的宿主细胞是内源存在细胞色素P450酶的底物或其底物前体的细胞。
本发明的又一目的是提供所述的表达构建物的用途,用于生产对-香豆酸。
本发明的又一目的是提供一种生产对-香豆酸的方法。所述方法包括:利用所述的细胞色素P450还原酶作为氧化还原分子伴侣,协助细胞色素P450酶CYP73A催化反式-肉桂酸合成对-香豆酸。
在一个优选例中,所述方法包括:以所述的表达构建物转化宿主细胞,利用转化的宿主细胞催化反式-肉桂酸合成对-香豆酸;所述的宿主细胞是原核细胞或真核细胞。常用的原核宿主细胞包括大肠杆菌、枯草杆菌、运动假单胞菌和乳酸菌等;常用的真核宿主细胞包括真菌细胞、植物细胞、昆虫细胞和哺乳动物细胞等。所述的真菌细胞包括酵母细胞。
在另一个优选例中,所述方法包括:以所述的表达构建物转化宿主细胞,培养转化的大肠杆菌细胞,利用简单的碳水化合物(葡萄糖)合成对-香豆酸。该宿主细胞是包含有细胞色素P450酶的底物的细胞;较佳地,该宿主细胞是内源存在反式-肉桂酸或其前体的细胞。
本发明首次揭示了石蒜科植物忽地笑来源的细胞色素P450还原酶LaCPR1,其具有良好的辅酶专一性。本发明还揭示了编码所述细胞色素P450还原酶的多核苷酸、表达所述细胞色素P450还原酶的表达载体及宿主细胞。本发明应用石蒜科植物来源的细胞色素P450还原酶,可协助细胞色素P450酶(CYPs)发挥催化活性,进而实现天然产物(例如但不限于对-香豆酸)的生物转化以及生物合成。
附图说明
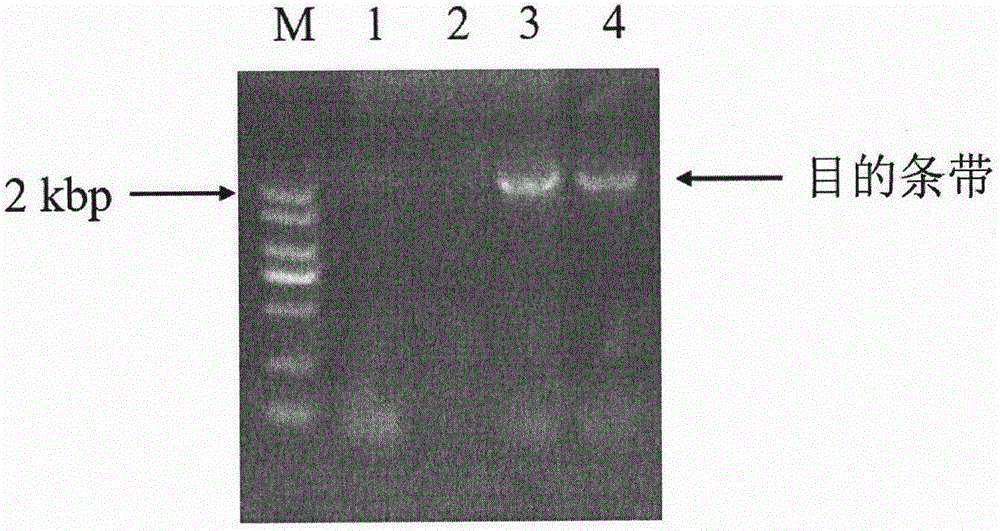
图1是引物对SEQ ID NO:3和SEQ ID NO:4的聚合酶链式反应(PCR)扩增产物的琼脂糖凝胶电泳检测结果。
图2是重组表达载体pET28a-LaCPR1的菌落PCR验证电泳图(引物为T7和T7ter)。
图3是重组表达载体pET28a-LaCPR1(ΔN58)的菌落PCR验证电泳图(引物为T7和T7ter)。
图4是LaCPR1在大肠杆菌中表达与分布检测的SDS-PAGE电泳图。
图5是LaCPR1的辅因子专一性与酶活性图。
图6是LaCPR1(ΔN58)在大肠杆菌中表达与分布检测的SDS-PAGE电泳图。
图7是LaCPR1(ΔN58)的辅因子专一性与酶活性图。
图8是重组表达载体pET28a-LaCPR1-CYP73A的菌落PCR验证电泳图(引物为SEQ ID NO:10和T7ter)。
图9是重组表达载体pET28a-LaCPR1(ΔN58)-CYP73A的菌落PCR验证电泳图(引物为SEQ ID NO:10和T7ter)。
图10是重组表达载体pET28a-CYP73A的菌落PCR验证电泳图(引物为T7和T7ter)。
图11是重组菌株EcR-LaCPR1、EcR-CYP73A、EcR-LaCPR1-CYP73A和EcR-LaCPR1(ΔN58)-CYP73A生物转化图。
图12是重组表达载体pET28a-LaCPR1(ΔN58)-CYP73A-rbsPAL的菌落PCR验证电泳图(引物为SEQ ID NO:13和T7ter)。
图13是重组菌株Ec31884-LaCPR1(ΔN58)-CYP73A-PAL发酵生产对-香豆酸的产量图。
具体实施方式
以下结合具体实施例并附图,进一步阐述本发明。
以下实施例进一步说明本发明的内容,但不应理解为对本发明的限制。在不背离本发明精神和实质的情况下,对本发明方法、步骤或条件所作的修改或替换,均属于本发明的范围。
若未特别指明,实施例中所用的技术手段为本领域技术人员所熟知的常规手段。
实施例1、细胞色素P450还原酶LaCPR1编码基因的克隆
合成两条引物分别具有序列表中SEQ ID NO:3、SEQ ID NO:4的核苷酸序列。
以从忽地笑中提取的RNA反转录获得的cDNA为模板,利用如上两条引物SEQ ID NO:3和SEQ ID NO:4进行PCR。DNA聚合酶选用南京诺唯赞生物科技有限公司的Super-Fidelity DNA聚合酶。PCR扩增程序为:95℃5min;94℃45s,56℃45s,72℃2min,共30个循环;72℃10min,降至10℃。PCR产物经琼脂糖凝胶电泳检测,结果如图1。
在紫外灯照射下,切下目标DNA条带。然后采用多功能DNA纯化试剂盒(离心柱型)(北京百泰克生物技术有限公司)从琼脂糖凝胶中回收DNA即为扩增出的细胞色素P450还原酶编码基因的DNA片段。利用宝生物工程(大连)有限公司(TaKaRa)的pMD19-T克隆试剂盒,将回收的PCR产物克隆到pMD19-T载体,所构建的载体命名为pMDT-LaCPR1。经测序获得LaCPR1的基因序列。
LaCPR1基因具有序列表中SEQ ID NO:2的核苷酸序列。自SEQ ID NO:2的5’-端第1-2088位核苷酸为LaCPR1的开放阅读框(Open Reading Frame,ORF),自SEQ ID NO:2的5’-端的第1-3位核苷酸为LaCPR1基因的起始密码子ATG,自SEQ ID NO:2的5’-端的第2086-2088位核苷酸为LaCPR1基因的终止密码子TGA。细胞色素P450还原酶编码基因LaCPR1编码一个含有695个氨基酸的蛋白质LaCPR1,具有SEQ ID NO:1的氨基酸序列,用软件预测到该蛋白质的理论分子量大小为77.4KDa,等电点pI为5.27。
实施例2、LaCPR1基因的重组表达载体的构建
(1)合成分别具有序列表中SEQ ID NO:5和SEQ ID NO:7核苷酸序列的两条引物。在合成的引物SEQ ID NO:5和SEQ ID NO:7的5’-端分别设置NdeI和XhoI两个酶切位点及其保护碱基序列,以忽地笑的cDNA为模板进行PCR扩增。PCR扩增程序同实施例1。PCR扩增产物经琼脂糖凝胶电泳检测、分离、切胶回收后经NdeI和XhoI双酶切,利用宝生物工程(大连)有限公司(TaKaRa)的T4DNA连接酶连接同样经NdeI和XhoI双酶切的pET28a载体(Novagen)中。连接产物转化大肠杆菌(E.coli)DH5α(购自南京诺唯赞生物科技有限公司)感受态细胞,并涂布于添加25μg/mL卡那霉素的LB平板上。通过菌落PCR验证获得阳性转化子,菌落PCR产物的琼脂糖电泳结果如图2。通过测序进一步验证重组质粒pET28a-NdeI-LaCPR1-XhoI构建成功,并在NdeI和XhoI酶切位点之间含有SEQ ID NO:2的全长多核苷酸序列。所获得的重组质粒命名为pET28a-LaCPR1。
(2)合成分别具有序列表中SEQ ID NO:6和SEQ ID NO:7核苷酸序列的两条引物。在合成的引物SEQ ID NO:6和SEQ ID NO:7的5’-端分别设置NdeI和XhoI两个酶切位点及其保护碱基序列,以忽地笑的cDNA为模板进行PCR扩增。PCR扩增程序同实施例1。PCR扩增产物经琼脂糖凝胶电泳检测、分离、切胶回收后经NdeI和XhoI双酶切,利用T4DNA连接酶(购自宝生物工程(大连)有限公司(TaKaRa))连接同样经NdeI和XhoI双酶切的pET28a载体(Novagen)中。连接产物转化大肠杆菌(E.coli)DH5α(购自南京诺唯赞生物科技有限公司)感受态细胞,并涂布于添加卡那霉素(终浓度为25μg/mL)的LB平板上。通过菌落PCR验证阳性转化子,菌落PCR产物的琼脂糖电泳结果如图3。测序进一步验证重组质粒pET28a-NdeI-LaCPR1(ΔN58)-XhoI构建成功,并在NdeI和XhoI酶切位点之间含有SEQ ID NO:2多核苷酸序列中175-2088位核苷酸。所获得的重组质粒命名为pET28a-LaCPR1(ΔN58)。
实施例3、LaCPR1和LaCPR1(ΔN58)的表达、纯化与酶活测定
(1)将重组质粒pET28a-LaCPR1利用热击法(42℃,90s)转化进入大肠杆菌Rosseta(DE3)感受态细胞中,获得重组大肠杆菌Rosseta(DE3)/pET28a-LaCPR1。挑取单克隆过夜培养,再将菌液按100倍稀释于含卡那霉素(终浓度为25μg/mL)的LB培养基中培养。待菌液生长至600nm波长下吸光度为0.6-0.8时,加入诱导剂异丙基-β-D-硫代半乳糖苷(IPTG)(终浓度为0.1mmol/L)进行诱导培养,培养温度为25℃。取诱导过的菌液40ml,离心收集菌体(4000rpm,10min,4℃),丢弃上清后用冰冷的1×PBS缓冲液洗涤菌体2遍,重悬于30ml 1×PBS缓冲液中。取1ml重悬菌液备用,剩余菌液进行超声破碎细胞,然后高速低温冷冻离心(12000g,30min,4℃)分离可溶部分和不可溶部分。取菌液、可溶部分和不可溶部分各400μl,分别加100μl 5×SDS Loading Buffer,短暂漩涡震荡后,于沸水中煮5min,10000rpm离心10min,各取10μl样品进行聚丙烯酰胺凝胶电泳(SDS-PAGE)检测重组蛋白,结果见图4,表达蛋白的分子量约为77KDa。取细菌破碎液上样到镍柱(购自南京金斯瑞生物科技有限公司),待滤液全部过滤完后,用Wash Buffer(20mM Tris-HCl,500mM NaCl,50mM imidazole,pH7.9)冲洗2次。然后用Elution Buffer(20mM Tris-HCl,500mM NaCl,250mM imidazole,pH7.9)洗脱。用Vivaspin浓缩洗脱液,并经PD-10脱盐柱(GE Healthcare)脱盐后,溶解于含10%甘油的Tris-HCl缓冲液中(50mM,pH8.0),进行聚丙烯酰胺凝胶电泳(SDS-PAGE)检测重组蛋白,并采用Bradford方法(Bradford et al.,1976)测定蛋白浓度,然后分装保存于-80℃备用。取纯化的LaCPR1蛋白液利用Guengerich等的方法(Guengerich et al.,Nature Protocol,2009)测定酶活,结果如图5,LaCPR1主要传递电子供体还原型烟酰胺腺嘌呤二核苷酸磷酸(NADPH)(还原力)中的电子。
(2)将重组质粒pET28a-LaCPR1(ΔN58)利用热击法(42℃,90s)转化进入大肠杆菌Rosseta(DE3)感受态细胞中,获得重组大肠杆菌Rosseta(DE3)/pET28a-LaCPR1(ΔN58)。挑取单克隆过夜培养,再将菌液按100倍稀释于含卡那霉素(终浓度为25μg/mL)的LB培养基中培养。待菌液生长至600nm波长下吸光度为0.6-0.8时,加入诱导剂异丙基-β-D-硫代半乳糖苷(IPTG)(终浓度为0.1mmol/L)进行诱导培养,培养温度为25℃。取诱导过的菌液40ml,离心收集菌体(4000rpm,10min,4℃),丢弃上清后用冰冷的1×PBS缓冲液洗涤菌体2遍,重悬于30ml 1×PBS缓冲液中。取1ml重悬菌液备用,剩余菌液进行超声破碎细胞,然后高速低温冷冻离心(12000g,30min,4℃)分离可溶部分和不可溶部分。取菌液、可溶部分和不可溶部分各400μl,分别加100μl 5×SDS Loading Buffer,短暂漩涡震荡后,于沸水中煮5min,10000rpm离心10min,各取10μl样品进行聚丙烯酰胺凝胶电泳(SDS-PAGE)检测重组蛋白,结果见图6,表达蛋白的分子量约为73KDa,其主要分布在大肠杆菌细胞破碎液的可溶性部分。取细菌破碎液上样到镍柱(购自南京金斯瑞生物科技有限公司),待滤液全部过滤完后,用Wash Buffer(20mM Tris-HCl,500mM NaCl,50mM imidazole,pH7.9)冲洗2次。然后用Elution Buffer(20mM Tris-HCl,500mM NaCl,250mM imidazole,pH7.9)洗脱。用Vivaspin浓缩洗脱液,并经PD-10脱盐柱(GE Healthcare)脱盐后,溶解于含10%甘油的Tris-HCl缓冲液中(50mM,pH8.0),进行聚丙烯酰胺凝胶电泳(SDS-PAGE)检测重组蛋白,并采用Bradford方法(Bradford et al.,1976)测定蛋白浓度,然后分装保存于-80℃备用。取纯化的LaCPR1(ΔN58)蛋白液利用Guengerich等的方法(Guengerich et al.,Nature Protocol,2009)测定酶活,结果如图7,LaCPR1(ΔN58)的酶活是LaCPR1的2.23倍。
实施例4、LaCPR1和CYP73A重组表达载体的构建
(1)合成分别具有序列表中SEQ ID NO:8和SEQ ID NO:9核苷酸序列的两条引物,以忽地笑的cDNA为模板进行PCR扩增CYP73A编码基因。PCR扩增程序同实施例1。PCR产物经琼脂糖凝胶电泳分离、回收。回收获得的DNA片段两端分别带有25bp与pET28a-LaCPR1载体XhoI酶切位点两侧同源的序列。利用XhoI限制性内切酶(购自宝生物工程(大连)有限公司(TaKaRa))将pET28a-LaCPR1载体线性化。利用One-step cloning试剂盒(购自南京诺唯赞生物科技有限公司)按照产品说明书将CYP73A编码基因导入pET28a-LaCPR1载体的XhoI酶切位点中,利用合成的具有序列表中SEQ ID NO:10核苷酸序列的引物与通用测序引物T7ter进行菌落PCR验证,获得重组表达质粒pET28a-LaCPR1-CYP73A,结果如图8。进一步测序证实重组质粒中LaCPR1和CYP73A进行了基因的融合表达。
(2)合成分别具有序列表中SEQ ID NO:8和SEQ ID NO:9核苷酸序列的两条引物,以忽地笑的cDNA为模板进行PCR扩增CYP73A编码基因。PCR扩增程序同实施例1。PCR产物经琼脂糖凝胶电泳分离、回收。回收获得的DNA片段两端分别带有25bp与pET28a-LaCPR1(ΔN58)载体XhoI酶切位点两侧同源的序列。利用XhoI限制性内切酶(购自宝生物工程(大连)有限公司(TaKaRa))将pET28a-LaCPR1(ΔN58)载体线性化。利用One-step cloning试剂盒(购自南京诺唯赞生物科技有限公司)按照产品说明书将CYP73A编码基因导入pET28a-LaCPR1(ΔN58)载体的XhoI酶切位点中,利用合成的具有序列表中SEQ ID NO:10核苷酸序列的引物与通用测序引物T7ter进行菌落PCR验证,获得重组表达质粒pET28a-LaCPR1(ΔN58)-CYP73A,结果如图9。进一步测序证实重组质粒中LaCPR1(ΔN58)和CYP73A进行了基因的融合表达。
(3)合成分别具有序列表中SEQ ID NO:11和SEQ ID NO:12核苷酸序列的两条引物,在合成的引物SEQ ID NO:11和SEQ ID NO:12的5’-端分别设置NdeI和XhoI两个酶切位点及其保护碱基序列,以忽地笑的cDNA为模板进行PCR扩增CYP73A编码基因。PCR扩增程序同实施例1。PCR扩增产物经琼脂糖凝胶电泳检测、分离、切胶回收后经NdeI和XhoI双酶切,利用T4DNA连接酶(购自宝生物工程(大连)有限公司(TaKaRa))连接同样经NdeI和XhoI双酶切的pET28a载体(Novagen)中。连接产物转化大肠杆菌(E.coli)DH5α(购自南京诺唯赞生物科技有限公司)感受态细胞,并涂布于添加卡那霉素(终浓度为25μg/mL)的LB平板上。利用通用测序引物对T7和T7ter进行菌落PCR验证阳性转化子,获得重组表达质粒pET28a-CYP73A,菌落PCR产物的琼脂糖电泳结果如图10。
实施例5、LaCPR1和LaCPR1(ΔN58)协同CYP73A催化反式-肉桂酸合成对-香豆酸
(1)配制培养基。诱导培养基(1L):31g Na2HPO4,15g KH2PO4,2.5g NaCl,5.0g NH4Cl,0.24g MgSO4,0.01g CaCl2,10g Glucose,1mL微量元素母液。生物转化培养基(1L):31g Na2HPO4,15g KH2PO4,2.5g NaCl,0.24g MgSO4,0.01g CaCl2,20g Glucose,1mL微量元素母液。微量元素母液配方为(1L):0.15mM(NH4)6Mo7O24,20.0mM H3BO3,1.5mM CoCl2,0.5mM CuSO4,4.0mM MnCl2,0.5mM ZnSO4,100mM HCl。
(2)将重组质粒pET28a-LaCPR1、pET28a-CYP73A、pET28a-LaCPR1-CYP73A和pET28a-LaCPR1(ΔN58)-CYP73A分别转化进入大肠杆菌Rosseta(DE3)细胞,获得重组菌株Rosseta(DE3)/pET28a-LaCPR1、Rosseta(DE3)/pET28a-CYP73A、Rosseta(DE3)/pET28a-LaCPR1-CYP73A和Rosseta(DE3)/pET28a-LaCPR1(ΔN58)-CYP73A,分别命名为菌株EcR-LaCPR1、EcR-CYP73A、EcR-LaCPR1-CYP73A和EcR-LaCPR1(ΔN58)-CYP73A。每个菌株各挑取单克隆接种添加卡那霉素(终浓度为25μg/ml)的LB培养基于37℃、200rpm过夜培养。
(3)将过夜培养菌液按100倍稀释于含卡那霉素(终浓度为25μg/mL)的50mL诱导培养基中培养。待菌液生长至600nm波长下吸光度为0.6-0.8时,加入诱导剂异丙基-β-D-硫代半乳糖苷(IPTG)(终浓度为0.1mmol/L)进行诱导培养,诱导时间为12h,诱导温度为25℃。
(4)收集诱导12h的细菌培养液于8000rpm、4℃离心5min,弃上清后用40mL生物转化培养基洗涤菌体1次。菌体用40ml生物转化培养基重悬后全部转移到250ml无菌三角瓶中,加入反式-肉桂酸(购自生工生物工程(上海)股份有限公司)二甲基亚砜(DMSO)母液至终浓度为100μM,于25℃、250rpm震荡培养60h。
(5)取生物转化液1ml于4℃冻融,加入等体积的甲醇,振荡混匀。室温下,12000rpm,离心5min。取上清用0.22μm孔径滤膜过滤,滤液上样高效液相色谱仪(HPLC)进行分析。分析条件为:采用LC-20A高效液相色谱仪(岛津,日本),InertSustain C18色谱柱(5μm,4.6mm×250mm),30℃柱温,二极管阵列检测器,278nm和314nm波长,10μl进样量,1.5%(v/v)乙酸水溶液为流动相A,100%乙腈为流动相B,0.9mL/min流速,梯度洗脱。分析结果如图11,结果表明:LaCPR1和CYP73A单独存在时并不能够催化底物反式-肉桂酸合成产物对-香豆酸;只有在LaCPR1或LaCPR1(ΔN58)的存在下,CYP73A才能够催化底物反式-肉桂酸合成产物对-香豆酸。可以理解为,LaCPR1和LaCPR1(ΔN58)具有细胞色素P450还原酶活性,可用于传递电子给细胞色素P450酶并协助细胞色素P450酶对底物的转化和产物的合成。
实施例6、重组大肠杆菌Ec31884-LaCPR1(ΔN58)-CYP73A-PAL的构建
(1)合成分别具有序列表中SEQ ID NO:13和SEQ ID NO:14核苷酸序列的两条引物,以忽地笑的cDNA为模板进行PCR扩增苯丙氨酸氨基裂解酶(PAL)编码基因。PCR扩增程序为:95℃ 5min;94℃ 45s,56℃ 45s,72℃3.5min,共30个循环;72℃ 10min,降至10℃。PCR产物经琼脂糖凝胶电泳分离、回收。回收获得的DNA片段两端分别带有25bp与pET28a-LaCPR1(ΔN58)-CYP73A载体XhoI酶切位点两端同源的序列,并且PAL基因起始密码子上游12bp处添加了核糖体结合位点(rbs)序列(AGGAG)。利用XhoI限制性内切酶(购自宝生物工程(大连)有限公司(TaKaRa))将pET28a-LaCPR1(ΔN58)-CYP73A载体线性化。利用One-step cloning试剂盒(购自南京诺唯赞生物科技有限公司)按照产品说明书将CYP73A编码基因导入pET28a-LaCPR1(ΔN58)-CYP73A载体的XhoI酶切位点中,利用合成的具有序列表中SEQ ID NO:13核苷酸序列的引物与通用测序引物T7ter进行菌落PCR验证,获得重组表达质粒pET28a-LaCPR1(ΔN58)-CYP73A-rbsPAL,结果如图12。进一步测序证实重组质粒中PAL与LaCPR1(ΔN58)-CYP73A形成了多顺反子基因结构。
(2)利用λDE3溶原化试剂盒(购自Novagen)按照试剂盒说明书操作,将λDE3原噬菌体插入到大肠杆菌苯丙氨酸生产菌株ATCC31884(购自ATCC)基因组中,获得溶原化的宿主菌株大肠杆菌ATCC31884(DE3)。
(3)将重组质粒pET28a-LaCPR1(ΔN58)-CYP73A-rbsPAL转化进入大肠杆菌ATCC31884(DE3)细胞,获得重组菌株ATCC31884(DE3)/pET28a-LaCPR1(ΔN58)-CYP73A-rbsPAL,命名为菌株Ec31884-LaCPR1(ΔN58)-CYP73A-PAL。
实施例7、重组大肠杆菌Ec31884-LaCPR1(ΔN58)-CYP73A-PAL发酵合成对-香豆酸
(1)配制发酵培养基(1L):31g Na2HPO4,15g KH2PO4,2.5g NaCl,5.0g NH4Cl,0.24g MgSO4,0.01g CaCl2,30g Glucose,1mL微量元素母液。微量元素母液配方同实施例5。
(2)挑取菌株Ec31884-LaCPR1(ΔN58)-CYP73A-PAL单克隆接种添加卡那霉素(终浓度为25μg/ml)的LB培养基于37℃、200rpm过夜培养。
(3)将过夜培养菌液按100倍稀释于含卡那霉素(终浓度为25μg/mL)的50mL发酵培养基中培养。待菌液生长至600nm波长下吸光度为0.6-0.8时,加入诱导剂异丙基-β-D-硫代半乳糖苷(IPTG)(终浓度为0.1mmol/L)进行诱导。培养温度转变为25℃,发酵时间为72h。
(4)取发酵液1ml于4℃冻融,加入等体积的甲醇,振荡混匀。室温下,12000rpm,离心5min。取上清用0.22μm孔径滤膜过滤,滤液上样高效液相色谱仪(HPLC)进行分析。分析条件同实施例5。分析结果如图13,结果表明:所构建的大肠杆菌菌株能够合成对-香豆酸,产物的量为400mg/L。可以理解为,基于能够合成CYP450酶的底物及其上游代谢产物的微生物菌株,LaCPR1(ΔN58)具有细胞色素P450还原酶活性,可用于传递电子给细胞色素P450酶并协助CYP450酶对底物的转化和产物的合成。
本发明提及的所有参考文献都在本申请中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述内容之后,本领域技术人员可以对本发明作各种改动或修饰,这些等价形式同样落于本申请所附权利要求书所限定的范围。
- 还没有人留言评论。精彩留言会获得点赞!
- 修饰细胞色素p450酶的编码核酸和其应用方法
- 一种改进细胞色素p450酶家族体外表达的方法
- 含有(e)-7-[4-(4-氟苯基)-6-异丙基-2-[甲基(甲基磺酰基)氨基]嘧啶-5基 ...的制作方法
- 含三氮杂螺[5.5]十一烷衍生物与细胞色素p450同工酶3a4抑制剂和/或p-糖蛋白抑制剂 ...的制作方法
- 细胞色素p450 bm-3 d168s变体酶以及应用其制备靛玉红的方法
- 细胞色素p450 bm-3 d168r变体酶以及应用其制备靛玉红的方法
- 用于检测人细胞色素p450相关的基因多态位点的引物系统及其用途的制作方法
- 人细胞色素p450 3a5酶及nadph-细胞色素p450氧化还原酶共表达体系的制作方法
- 一种检测黄鳝芳香化酶p450基因等位基因甲基化状态的方法
- 四翅滨藜细胞色素p450基因克隆及其应用的制作方法