呋喃卡山烷二萜衍生物及其药物组合物和其在制药中的应用的制作方法
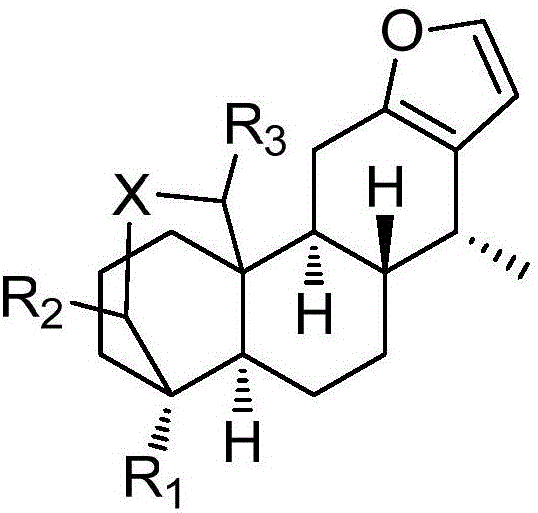
本发明属于药物技术领域,具体地,涉及呋喃卡山烷二萜衍生物,以其为药物有效成分的药物组合物,它们在制备预防和治疗2型糖尿病及代谢性疾病的药物中的应用。
背景技术:
糖尿病(diabetes mellitus,DM)是由胰岛素缺失和(或)胰岛素抵抗引发的慢性代谢疾病。近年来,糖尿病发病率一直呈现上升趋势,根据权威机构统计(国际糖尿病联盟IDF,2015年数据),2015年全球糖尿病患者达4.15亿,其中80%以上分布于发展中国家,预计2040年此数字将达6.42亿。随着生活水平提高和生活方式改变,中国糖尿病患病率急剧上升,数据也显示,2015年中国糖尿病患病人数高达1.09亿,患病率接近10%,居世界首位,已成为严重危害我国公民健康的慢性疾病。糖尿病已成为继心脑血管疾病和肿瘤之后第三大非传染性疾病,是目前严重危害人类健康的主要疾病之一。
糖尿病主要分为两类,1型糖尿病(胰岛素依赖型糖尿病,IDDM)和2型糖尿病(非胰岛素依赖型糖尿病,NIDDM)。其中,2型糖尿病约占糖尿病总体人群的90‐95%以上。目前用于治疗2型糖尿病的口服药物主要有胰岛素增敏剂(噻唑烷二酮类)、促进胰岛素分泌药(磺酰脲和非磺酰脲类)、双胍类药及α‐葡萄糖苷酶抑制剂。此外,DPPIV抑制剂类药物、GLP‐1类似物类药物、SGLT2抑制剂类药物,是近10年来上市使用的新型抗糖尿病药物。
研究表明,导致糖尿病的主要病理基础是胰岛素分泌不足、胰岛素抵抗和内源葡萄糖输出增加,从而引起糖尿病病人餐前和餐后高血糖。目前临床上应用治疗糖尿病的药物主要是针对胰岛素分泌不足和改善胰岛素抵抗。二甲双胍(metformin)是目前临床唯一用于非直接性减少内源性葡萄糖输出的药物。已有研究表明,内源性葡萄糖生成增加是造成糖尿病患者空腹血糖升高的主要原因。内源性葡萄糖主要来源于肝脏,肝脏产生葡萄糖主要是通过内源性合成葡萄糖,即糖异生作用。因此,有效抑制肝脏过度糖异生,减少内源性葡萄糖生成,对于维持正常血糖水平具有重要意义,是治疗2型糖尿病的重要靶标之一。
药用植物是发现抗糖尿病活性成分的重要资源,近年来从中发现结构多样活性成分,包括黄酮、木脂素、二萜、三萜类化合物等,有望开发在临床上应用。卡山烷二萜类化合物具有广泛的生物活性,如抗肿瘤、抗菌、抗炎、抗疟疾等功能(Wu,et.al.,ChemistryBiodiversity,2011,8,1370‐1399)。但目前尚无呋喃卡山烷二萜类化合物在治疗和预防糖尿病及代谢性疾病等方面的报道。
技术实现要素:
本发明的目的在于:提供一类新的呋喃卡山烷二萜衍生物和phangininE及其衍生物,以其为活性成分的药物组合物,其制备方法,以及该类化合物及其药物组合物在制备预防和治疗2型糖尿病及代谢性疾病的药物中的应用。
为了实现本发明的上述目的,本发明提供了如下的技术方案:
式(I)所示的呋喃卡山烷二萜衍生物,
式中,X选自O或NH;
R1选自氢、甲基、醛基、羧基、羟甲基(‐CH2OH)、亚甲基卤素(卤素为氟、氯、溴)、C1‐10烷氧基亚甲基、C2‐10酰氧基亚甲基、‐COOR,其中R为C2‐10烷基、异氰酸酯基、‐NHCONR2,其中R为H和C1‐10烷基或H和C2‐10酰基或都为C1‐10烷基、‐CH2NH2/‐CH2NR2,其中R为H和C1‐10烷基或H和C2‐10酰基或都为C1‐10烷基;
R2、R3分别选自H,羰基、C1‐10烷氧基;
如式(I)所示的呋喃卡山烷二萜衍生物,为如下式(II)所示的呋喃半日花烷二萜衍生物,
式中,X选自O或NH;
R选自氢、甲基、醛基、羧基、羟甲基(‐CH2OH)、亚甲基卤素(卤素为氟、氯、溴)、C1‐10烷氧基亚甲基、C2‐10酰氧基亚甲基、‐COOR,其中R为C2‐10烷基、异氰酸酯基、‐NHCONR2,其中R为H和C1‐10烷基或H和C2‐10酰基或都为C1‐10烷基、‐CH2NH2/‐CH2NR2,其中R为H和C1‐10烷基或H和C2‐10酰基或都为C1‐10烷基;
如式(I)所示的呋喃卡山烷二萜衍生物,为如下结构式(Ⅳ)和式(Ⅴ)所示的卡山烷二萜衍生物,
式中,R选自氢、甲基、醛基、羧基、羟甲基(‐CH2OH)、亚甲基卤素(卤素为氟、氯、溴)、C1‐10烷氧基亚甲基、C2‐10酰氧基亚甲基、‐COOR,其中R为C2‐10烷基、异氰酸酯基、‐NHCONR2,其中R为H和C1‐10烷基或H和C2‐10酰基或都为C1‐10烷基、‐CH2NH2/‐CH2NR2,其中R为H和C1‐10烷基或H和C2‐10酰基或都为C1‐10烷基;
如上所示的呋喃半日花烷二萜,为如下化合物:
下述式(Ⅵ)所示的呋喃卡山烷二萜phangininE及其衍生物及其为活性成分的药物组合物在制备治疗或预防2型糖尿病或代谢性疾病的药物中的应用,
其中,R1和R2选自氢、羟基、甲氧基或羰基,当R1为羰基,R2为氢原子时,化合物为phanginin E;当R2为羰基,R1为氢原子时,化合物为19‐deoxo‐20‐oxophanginin E。
本发明提供治疗或预防糖尿病及代谢性疾病的药物组合物,其中含有治疗有效量的上述的呋喃卡山烷二萜衍生物,及可药用载体。
本发明提供了上述呋喃卡山烷二萜衍生物或其药物组合物在制备治疗或预防2型糖尿病的药物中的应用。
本发明提供了上述呋喃卡山烷二萜衍生物或其药物组合物在制备治疗或预防代谢性疾病的药物中的应用。
本发明提供了phanginin E在治疗或预防2型糖尿病或代谢性疾病的药物中的应用。
本发明同时提供了19‐deoxo‐20‐oxophanginin E在治疗或预防2型糖尿病或代谢性疾病的药物中的应用。
本发明另外还提供了所述的呋喃卡山烷二萜化合物1‐13的制备方法,该方法包括下述步骤:
化合物P1和P2的制备:
苏木种子10kg,粉碎后用95%乙醇于室温浸泡提取三次,每次48小时,合并提取液减压浓缩得粗提物,将粗提物分散于水中,用等体积的乙酸乙酯萃取四次,减压浓缩萃取液,乙酸乙酯萃取物经硅胶柱层析反复柱层析及重结晶得到化合物P1和P2;
化合物1和2的制备:
化合物P1溶于10mL甲醇溶剂中,加入对甲苯磺酸,于室温反应8h,向反应液中水15mL,用乙酸乙酯萃取,有机相用饱和NaCl洗涤,无水硫酸钠干燥,减压浓缩得淡黄色粉末,经硅胶柱层析石油醚:丙酮=8:2得到化合物1和化合物2;
化合物3和4的制备:
化合物P2溶于20mL甲醇溶剂中,加入对甲苯磺酸64mg,于室温反应8h,向反应液中水20mL,用乙酸乙酯萃取,有机相用饱和NaCl洗涤,无水硫酸钠干燥,减压浓缩得淡黄色粉末。经硅胶柱层析石油醚:丙酮=8:2得到化合物3和化合物4;
化合物5的制备:
化合物P1溶于2mL二氯甲烷溶剂中,加入PDC31mg和4A分子筛31mg,于室温反应过夜,反应液用硅藻土过滤,减压浓缩蒸干,得淡黄色固体,经硅胶柱层析石油醚:乙酸乙酯=8:2得到化合物5;
化合物6的制备:
化合物P2溶于3mL二氯甲烷溶剂中,加入PDC90mg和4A分子筛90mg,于室温反应过夜,反应液用硅藻土过滤,减压浓缩蒸干,得淡黄色固体。经硅胶柱层析石油醚:乙酸乙酯=8:2得到化合物6;
化合物7的制备:
化合物1溶于9mL四氢呋喃溶剂中,加入LiAlH440mg于75℃下加热反应4h,向反应液中加入1mL 5%硫酸溶液搅拌5分钟,用乙酸乙酯萃取,萃取液用饱和碳酸氢钠溶液洗涤3次,无水硫酸钠干燥,减压浓缩得淡黄色固体。经硅胶柱层析石油醚:乙酸乙酯=9:1得到化合物7;
化合物8的制备:
化合物7溶于5mL二氯甲烷溶剂中,在冰浴条件下加入乙酸酐200μL、三乙胺200μL、DMAP 3.0mg,随后自然升到室温反应2h,反应液加水稀释,水相用乙酸乙酯萃取,萃取液用饱和氯化钠溶液洗涤3次,无水硫酸钠干燥,减压浓缩得淡黄色固体,经硅胶柱层析石油醚:乙酸乙酯=20:1得到化合物8;
化合物9的制备:
化合物4溶于6mL四氢呋喃溶剂中,加入LiAlH430mg于75℃下加热反应4h,向反应液中加入1mL 5%硫酸溶液搅拌5分钟,用乙酸乙酯萃取,萃取液用饱和碳酸氢钠溶液洗涤3次,无水硫酸钠干燥,减压浓缩得淡黄色固体,经硅胶柱层析石油醚:乙酸乙酯=9:1得到化合物9;
化合物10的制备:
将化合物9溶于5mL二氯甲烷溶剂中,在冰浴条件下加入乙酸酐100μL、三乙胺100μL、DMAP1.5mg,随后自然升到室温反应2h,反应液加水稀释,水相用乙酸乙酯萃取,萃取液用饱和氯化钠溶液洗涤3次,无水硫酸钠干燥,减压浓缩得淡黄色固体,经硅胶柱层析石油醚:乙酸乙酯=20:1得到化合物10;
化合物11的制备:
化合物P2溶于6mL四氢呋喃溶剂中,加入LiAlH430mg于75℃下加热反应4h,向反应液中加入1mL 5%硫酸溶液搅拌5分钟,用乙酸乙酯萃取,萃取液用饱和碳酸氢钠溶液洗涤3次,无水硫酸钠干燥,减压浓缩,浓缩后的样品继续溶于5mL二氯甲烷溶剂中,加入IBX 300mg于室温反应12h,加入适量饱和碳酸氢钠溶液,用乙酸乙酯萃取3次,水硫酸钠干燥,减压浓缩得淡黄色固体,经硅胶柱层析石油醚:乙酸乙酯=8:2得到化合物11;
化合物12的制备:
化合物9溶于2mL二氯甲烷溶剂中,加入IBX 100mg于室温反应12h,加入适量饱和碳酸氢钠溶液,用乙酸乙酯萃取3次,水硫酸钠干燥,减压浓缩得淡黄色固体,经硅胶柱层析石油醚:乙酸乙酯=8:2得到化合物12;
化合物13的制备:
化合物P1 20mg溶于6mL四氢呋喃溶剂中,加入KOH10mg于75℃下加热反应3h,冷却至室温,向反应液中加入1mL 1N盐酸溶液搅拌5分钟,用乙酸乙酯萃取,萃取液用饱和碳酸氢钠溶液洗涤3次,无水硫酸钠干燥,减压浓缩,浓缩后的样品继续溶于5mL二氯甲烷溶剂中,加入PDC 30mg于室温反应12h,加入适量饱和碳酸氢钠溶液,用乙酸乙酯萃取3次,水硫酸钠干燥,减压浓缩得淡黄色固体,经硅胶柱层析石油醚:乙酸乙酯=8:2得到化合物13。
本发明中的化合物用作药物时,可以直接使用,或者以药物组合物的形式使用。也可以与其他药物组成复方的形式使用,该药物组合物含有0.1~99%,优选为0.5~90%的本发明化合物,其余为药物学上可接受的,对人和动物无毒和惰性的药物制剂常用的药用载体和/或赋形剂。将本发明的药物组合物以单位体重服用量的形式使用。可以使用不同的药用辅料,制成固体制剂(片剂、胶囊剂、颗粒剂、散剂等)。
除非另有说明,本发明使用的术语“烷基”是指直链或者支链的饱和一价烃基,其中烷基可以任选地被一个或多个取代基取代。本发明使用的直链C1‐6和支链C3‐6烷基也被称为“低级烷基”。烷基的实施方式包括但不限于甲基、乙基、丙基(包括所有同分异构形式)、n‐丙基、异丙基、丁基(包括所有同分异构形式)、n‐丁基、异丁基、t‐丁基、戊基(包括所有同分异构形式)、和己基(包括所有同分异构形式)。例如,C1‐6烷基指具有1到6个碳原子的直链饱和一价烃基或者具有3到6个碳原子的支链饱和一价烃基。
除非另有说明,本发明使用的术语“酰基”是指直链或支链,环状或非环状的饱和烃基取代的碳酰或磺酰基,也可以是烯基、炔基、芳基等不饱和烃基取代的碳酰或磺酰基。
除非另作说明,本发明使用的术语“烯基”是指包含一个或多个碳碳双键的直链或者支链一价烃基。烯基可以任选地被一个或多个取代基取代。术语“烯基”也包括“顺式(cis)”和“反式(trans)”结构的基团,或者本领域普通技术人员所理解的“E”和“Z”式结构。除非另作说明,本发明使用的术语“烯基”包括直链和支链烯基。例如,C2‐6烯基是指2到6个碳原子的直链不饱和一价烃基或者3到6个碳原子的支链不饱和一价烃基。
除非另作说明,本发明使用的术语“炔基”是指包含一个或多个碳碳三键的直链或者支链一价烃基。炔基可以任选地被一个或多个取代基取代。除非另作说明,术语“炔基”也包括直链和支链炔基。
除非另作说明,本发明使用的术语“芳基”是指单环芳基和/或包含至少一个芳香烃环的多环单价芳基。
除非另作说明,本发明使用的术语“杂烷基”、“杂烯基”和“杂炔基”分别是指一个或多个碳原子被杂原子替换的烷基、链烯基和炔基。
除非另作说明,本发明使用的术语“杂原子”是指除了碳或者氢以外的其他任何原子。通常指N、O、S、Si或P。
附图说明
图1为本发明的呋喃卡山烷二萜衍生物对肝细胞糖异生抑制作用的活性结果图。
图2为本发明的呋喃卡山烷二萜衍生物结构示意图。
具体实施方式
以下结合附图,通过本发明的实施例来进一步说明本发明的实质性内容,但并不以任何形式来限制本发明。
实施例1:
本发明的具体实验方法:
化合物P1和P2是从采自广西百色产豆科云石属植物苏木(Caesalpiniasappan)乙醇提取物中分离得到的。
化合物P1和P2的制备:
苏木(C.sappan)种子10kg,粉碎后用95%乙醇于室温浸泡提取三次,每次48小时,合并提取液减压浓缩得粗提物(450g)。将粗提物分散于水中,用等体积的乙酸乙酯萃取四次,减压浓缩萃取液。乙酸乙酯萃取物(200g)经硅胶柱层析反复柱层析及重结晶得到化合物P1(15g)和P2(6g)。
实施例2:
化合物1和2的制备
化合物P1(100mg)溶于10mL甲醇溶剂中,加入对甲苯磺酸(32mg),于室温反应8h。向反应液中水15mL,用乙酸乙酯萃取,有机相用饱和NaCl洗涤,无水硫酸钠干燥,减压浓缩得淡黄色粉末。经硅胶柱层析(石油醚:丙酮=8:2)得到化合物1(70mg,产率67%)和化合物2(12mg,产率11%)。
化合物1:白色粉末;1H NMR(500MHz,CDCl3)δ:7.22(1H,br s),6.20(1H,br s),4.41(1H,br s),4.17(1H,dd,J=11.7,2.5Hz),3.69(1H,dd,J=11.7,1.3Hz),3.67(3H,s),3.27(3H,s),2.72(1H,dd,J=15.4,5.1Hz),2.58(1H,m),2.23‐2.40(3H,m),2.09(1H,m),1.68‐2.08(3H,m),1.66(1H,m),1.53‐1.62(2H,m),1.45(1H,td,J=11.7,5.1Hz),1.36(1H,m),1.24(1H,m),1.16(1H,m),0.96(3H,d,J=7.1Hz).13C NMR(125MHz,CDCl3)δ:175.8(s),149.5(s),140.4(d),122.5(s),109.8(d),103.9(d),61.7(t),54.6(q),51.5(q),45.5(s),45.5(d),42.3(d),38.9(s),38.0(t),36.8(d),35.5(t),31.5(d),29.5(t),23.5(t),22.5(t),21.1(t),16.9(q).正离子ESIMS m/z397[M+Na]+;HRESIMS m/z397.1988[M+Na]+(calcd for C22H30O5Na,397.1991).
化合物2:白色粉末;1H NMR(500MHz,CDCl3)δ:7.22(1H,br s),6.17(1H,br s),4.81(1H,br s),4.29(1H,dd,J=12.1,1.6Hz),3.54(1H,d,J=12.1Hz),3.68(3H,s),3.38(3H,s),2.81(1H,dd,J=17.3,10.7Hz),2.62‐2.72(2H,m),2.42(1H,dd,J=13.0,4.8Hz),2.24(1H,m),1.89(1H,m),1.67‐1.82(4H,m),1.57(1H,m),1.51(1H,m),1.20‐1.48(3H,m),0.97(1H,m),0.95(3H,d,J=7.0Hz).13C NMR(125MHz,CDCl3)δ:175.9(s),150.1(s),140.3(d),121.4(s),109.5(d),101.8(d),65.1(t),55.8(q),51.7(q),47.0(d),46.1(s),43.0(d),39.9(s),36.4(d),35.9(t),33.4(t),31.6(d),30.1(t),23.7(t),23.0(t),20.8(t),17.6(q).正离子ESIMS m/z397[M+Na]+;HRESIMS m/z397.1989[M+Na]+(calcd for C22H30O5Na,397.1991).
实施例3:
化合物3和4的制备
化合物P2(200mg)溶于20mL甲醇溶剂中,加入对甲苯磺酸(64mg),于室温反应8h。向反应液中水20mL,用乙酸乙酯萃取,有机相用饱和NaCl洗涤,无水硫酸钠干燥,减压浓缩得淡黄色粉末。经硅胶柱层析(石油醚:丙酮=8:2)得到化合物3(114mg,产率55%)和化合物4(73mg,产率35%)。
化合物3:白色粉末;1H NMR(500MHz,CDCl3)δ:7.21(1H,d,J=1.4Hz),6.17(1H,d,J=1.4Hz),4.76(1H,br s),4.04(1H,dd,J=11.2,2.7Hz),3.70(3H,s),3.54(1H,d,J=11.2Hz),3.35(3H,s),2.71(1H,dd,J=16.1,5.9Hz),2.62(1H,m),2.16‐2.34(3H,m),2.06(1H,m),1.82‐1.93(2H,m),1.53‐1.73(5H,m),1.52(1H,J=11.6,5.9Hz),1.19‐1.36(2H,m),0.96(3H,d,J=7.1Hz).13C NMR(125MHz,CDCl3)δ:174.9(s),149.1(s),140.5(d),122.3(s),109.6(d),102.6(d),62.7(t),55.6(q),51.6(q),49.7(s),44.2(d),42.0(d),38.3(t),37.0(d),36.2(t),36.0(s),31.6(d),30.6(t),23.8(t),22.5(t),21.1(t),17.2(q).正离子ESIMS m/z397[M+Na]+;HRESIMS m/z397.1988[M+Na]+(calcd for C22H30O5Na,397.1991).
化合物4:白色粉末;1H NMR(500MHz,CDCl3)δ:7.21(1H,br s),6.17(1H,br s),5.05(1H,br s),4.03(1H,dd,J=11.6,2.7Hz),3.81(1H,dd,J=11.6,1.1Hz),3.69(3H,s),3.53(3H,s),2.72(1H,dd,J=16.3,6.0Hz),2.63(1H,m),2.31(1H,m),2.13‐2.26(3H,m),1.85(1H,m),1.75(1H,m),1.62‐1.74(3H,m),1.49‐1.63(2H,m),1.23‐1.37(3H,m),0.95(3H,d,J=7.0Hz).13C NMR(125MHz,CDCl3)δ:174.9(s),148.9(s),140.7(d),122.1(s),109.5(d),101.2(d),66.9(t),57.4(q),51.6(q),49.4(s),47.9(d),41.3(d),38.4(t),36.3(d),35.4(s),31.5(d),30.0(t),29.0(t),23.1(t),22.5(t),21.0(t),17.1(q).正离子ESIMS m/z397[M+Na]+;HRESIMS m/z397.1987[M+Na]+(calcd for C22H30O5Na,397.1991).
实施例4:
化合物5的制备
化合物P1(20mg)溶于2mL二氯甲烷溶剂中,加入PDC(31mg)和4A分子筛(31mg),于室温反应过夜。反应液用硅藻土过滤,减压浓缩蒸干,得淡黄色固体。经硅胶柱层析(石油醚:乙酸乙酯=8:2)得到化合物5(12mg,产率60%)。
化合物5:无色针晶(甲醇);1H NMR(500MHz,CDCl3)δ:7.21(1H,br s),6.16(1H,br s),4.78(1H,dd,J=12.5,1.7Hz),4.30(1H,d J=12.5Hz),3.74(1H,m),3.73(3H,s),3.27(3H,s),2.64‐2.74(2H,m),2.31(1H,br d,J=13.5Hz),1.87‐2.00(4H,m),1.55‐1.86(6H,m),1.42‐1.52(2H,m),1.31(1H,m),0.97(3H,d,J=7.1Hz).13C NMR(125MHz,CDCl3)δ:174.0(s),172.9(s),149.2(s),140.4(d),121.7(s),109.4(d),71.7(t),52.4(q),47.5(s),45.5(d),44.9(s),43.2(d),36.8(t),36.2(d),34.4(t),31.3(d),29.9(t),26.6(t),23.3(t),19.5(t),18.0(q).正离子ESIMS m/z381[M+Na]+;HRESIMS m/z381.1677[M+Na]+(calcd for C21H26O5Na,381.1678).
实施例5:
化合物6的制备
化合物P2(30mg)溶于3mL二氯甲烷溶剂中,加入PDC(90mg)和4A分子筛90mg),于室温反应过夜。反应液用硅藻土过滤,减压浓缩蒸干,得淡黄色固体。经硅胶柱层析(石油醚:乙酸乙酯=8:2)得到化合物6(27mg,产率90%)。
化合物6:无色针晶(甲醇);1H NMR(500MHz,CDCl3)δ:7.22(1H,br s),6.18(1H,br s),4.66(1H,dd,J=11.9,2.3Hz),4.24(1H,d,J=11.9Hz),3.77(3H,s),2.78(1H,dd,J=16.2,6.0Hz),2.70(1H,m),2.03‐2.20(4H,m),1.67‐1.92(5H,m),1.53‐1.64(2H,m),1.29‐1.49(3H,m),0.98(3H,d,J=7.1Hz).13C NMR(125MHz,CDCl3)δ:171.2(s),169.8(s),147.8(s),141.0(d),122.5(s),109.5(d),73.6(t),55.9(s),52.3(q),46.7(d),40.9(d),38.1(t),36.0(d),35.4(s),34.8(t),31.2(d),29.1(t),25.2(t),22.5(t),20.0(t),17.2(q).正离子ESIMS m/z381[M+Na]+;HRESIMS m/z381.1679[M+Na]+(calcd for C21H26O5Na,381.1678).
实施例6:
化合物7的制备
化合物1(40mg)溶于9mL四氢呋喃溶剂中,加入LiAlH4(40mg)于75℃下加热反应4h。向反应液中加入1mL 5%硫酸溶液搅拌5分钟,用乙酸乙酯萃取,萃取液用饱和碳酸氢钠溶液洗涤3次,无水硫酸钠干燥,减压浓缩得淡黄色固体。经硅胶柱层析(石油醚:乙酸乙酯=9:1)得到化合物7(33mg,产率90%)。
化合物7:白色粉末;1H NMR(500MHz,CDCl3)δ:7.22(1H,br s),6.20(1H,br s),4.45(1H,br s),3.76(1H,dd,J=11.0,2.3Hz),3.40,(1H,d,J=10.8Hz),3.37(1H,d,J=11.0,1.2Hz),3.29(1H,d,J=10.8Hz),3.26(3H,s),2.72(1H,dd,J=15.4,5.1Hz),2.58(1H,m),2.22‐2.40(3H,m),1.93‐2.07(2H,m),1.79(1H,dd,J=11.9,6.2Hz),1.55‐1.73(3H,m),1.12‐1.36(7H,m),0.95(3H,d,J=7.1Hz).13C NMR(125MHz,CDCl3)δ:149.8(s),140.3(d),122.4(s),109.8(d),104.7(d),67.4(t),63.8(t),54.5(q),44.7(d),42.5(d),39.1(s),38.5(t),37.4(s),36.5(d),34.4(t),31.5(t),29.7(t),22.6(t),21.4(t),21.2(t),16.9(q).正离子ESIMS m/z369[M+Na]+;HRESIMS m/z369.2030[M+Na]+(calcd for C21H30O4Na,369.2042).
实施例7:
化合物8的制备
化合物7(20mg)溶于5mL二氯甲烷溶剂中,在冰浴条件下加入乙酸酐(200μL)、三乙胺(200μL)、DMAP(3.0mg),随后自然升到室温反应2h。反应液加水稀释,水相用乙酸乙酯萃取,萃取液用饱和氯化钠溶液洗涤3次,无水硫酸钠干燥,减压浓缩得淡黄色固体。经硅胶柱层析(石油醚:乙酸乙酯=20:1)得到化合物8(18mg,产率80%)。
化合物8:无色晶体(甲醇);1H NMR(500MHz,CDCl3)δ:7.22(1H,br s),6.20(1H,br s),4.44(1H,br s),3.71‐3.83(3H,m),3.41(1H,m),3.26(3H,s),2.72(1H,dd,J=15.4,5.1Hz),2.58(1H,m),2.21‐2.39(3H,m),2.07(3H,s),1.93‐2.07(2H,m),1.84(1H,dd,J=13.1,6.1Hz),1.69(1H,m),1.39‐1.60(4H,m),1.11‐1.33(3H,m),0.96(3H,d,J=7.1Hz).13C NMR(125MHz,CDCl3)δ:171.3(s),149.7(s),140.3(d),122.4(s),109.8(d),104.4(d),68.9(t),63.4(t),54.6(q),45.3(d),42.5(d),39.1(s),38.4(t),36.4(d),36.1(s),34.8(d),31.5(d),29.6(t),22.5(t),21.6(t),21.1(t),20.9(q),16.9(q).正离子ESIMS m/z411[M+Na]+;HRESIMS m/z411.2142[M+Na]+(calcd for C23H32O5Na,411.2147).
实施例8:
化合物9的制备
化合物4(30mg)溶于6mL四氢呋喃溶剂中,加入LiAlH4(30mg)于75℃下加热反应4h。向反应液中加入1mL 5%硫酸溶液搅拌5分钟,用乙酸乙酯萃取,萃取液用饱和碳酸氢钠溶液洗涤3次,无水硫酸钠干燥,减压浓缩得淡黄色固体。经硅胶柱层析(石油醚:乙酸乙酯=9:1)得到化合物9(25mg,产率93%)。
化合物9:白色粉末;1H NMR(500MHz,CDCl3)δ:7.22(1H,br s),6.17(1H,br s),4.65(1H,br s),4.02(1H,dd,J=11.6,2.7Hz),3.83,(1H,d,J=11.6Hz),3.70(1H,d,J=11.0Hz),3.48(3H,s),3.22(1H,d,J=11.0Hz),2.72(1H,dd,J=16.3,6.0Hz),2.63(1H,m),2.14‐2.36(4H,m),1.48‐1.76(6H,m),1.17‐1.41(5H,m),0.95(3H,d,J=7.1Hz).13C NMR(125MHz,CDCl3)δ:149.1(s),140.1(d),122.1(s),109.6(d),105.0(d),68.6(t),67.3(t),56.7(q),47.8(d),41.3(d),40.0(s),39.1(t),36.2(d),35.9(s),31.6(d),30.2(t),29.0(t),22.5(t),21.2(t),20.5(t),17.2(q).正离子ESIMS m/z369[M+Na]+;HRESIMS m/z369.2030[M+Na]+(calcd for C21H30O4Na,369.2042).
实施例9:
化合物10的制备
化合物9(10mg)溶于5mL二氯甲烷溶剂中,在冰浴条件下加入乙酸酐(100μL)、三乙胺(100μL)、DMAP(1.5mg),随后自然升到室温反应2h。反应液加水稀释,水相用乙酸乙酯萃取,萃取液用饱和氯化钠溶液洗涤3次,无水硫酸钠干燥,减压浓缩得淡黄色固体。经硅胶柱层析(石油醚:乙酸乙酯=20:1)得到化合物10(9mg,产率81%)。
化合物10:白色粉末;1H NMR(500MHz,CDCl3)δ:7.21(1H,br s),6.17(1H,br s),4.52(1H,br s),4.02(1H,dd,J=11.5,2.6Hz),3.92,(1H,d,J=11.3Hz),3.89(1H,d,J=11.3Hz),3.82(1H,d,J=11.5Hz),3.41(3H,s),2.72(1H,dd,J=16.3,6.0Hz),2.64(1H,m),2.34(1H,m),2.14‐2.26(2H,m),2.09(3H,s),1.95(1H,dd,J=13.3,5.6Hz),1.46‐1.76(6H,m),1.17‐1.44(4H,m),0.96(3H,d,J=7.1Hz).13C NMR(125MHz,CDCl3)δ:171.3(s),149.1(s),140.6(d),122.1(s),109.6(d),102.8(d),67.6(t),67.3(t),57.0(q),46.0(d),41.3(d),39.5(s),38.8(t),36.4(d),35.9(s),31.6(d),30.3(t),29.4(t),22.5(t),21.1(t),21.0(q),20.7(t),17.2(q).正离子ESIMS m/z411[M+Na]+;HRESIMS m/z411.2144[M+Na]+(calcd for C23H32O5Na,411.2147).
实施例10:
化合物11的制备
化合物P2(30mg)溶于6mL四氢呋喃溶剂中,加入LiAlH4(30mg)于75℃下加热反应4h。向反应液中加入1mL 5%硫酸溶液搅拌5分钟,用乙酸乙酯萃取,萃取液用饱和碳酸氢钠溶液洗涤3次,无水硫酸钠干燥,减压浓缩。浓缩后的样品继续溶于5mL二氯甲烷溶剂中,加入IBX(300mg)于室温反应12h。加入适量饱和碳酸氢钠溶液,用乙酸乙酯萃取3次,水硫酸钠干燥,减压浓缩得淡黄色固体。经硅胶柱层析(石油醚:乙酸乙酯=8:2)得到化合物11(9mg,产率34%)。
化合物11:白色粉末;1H NMR(500MHz,CDCl3)δ:10.2(1H,s),7.22(1H,br s),6.18(1H,br s),4.66(1H,dd,J=12.0,2.2Hz),4.28,(1H,d,J=12.0Hz),2.78(1H,dd,J=16.1,6.0Hz),2.69(1H,m),2.16(1H,m),1.99‐2.12(2H,m),1.81‐1.94(3H,m),1.61‐1.79(4H,m),1.11(1H,m),1.21‐1.38(3H,m),0.95(3H,d,J=7.1Hz).13C NMR(125MHz,CDCl3)δ:202.1(d),172.0(s),147.4(s),141.0(d),122.6(s),109.5(d),73.9(t),56.8(s),44.6(d),40.4(d),37.9(t),36.3(t),35.5(s),33.7(t),31.3(d),28.9(t),24.3(t),22.5(t),21.2(t),20.0(t),17.3(q).正离子ESIMS m/z351[M+Na]+;HRESIMS m/z351.1575[M+Na]+(calcd for C20H24O4Na,351.1572).
实施例11:
化合物12的制备
化合物9(10mg)溶于2mL二氯甲烷溶剂中,加入IBX(100mg)于室温反应12h。加入适量饱和碳酸氢钠溶液,用乙酸乙酯萃取3次,水硫酸钠干燥,减压浓缩得淡黄色固体。经硅胶柱层析(石油醚:乙酸乙酯=8:2)得到化合物12(9mg,产率90%)。
化合物12:白色粉末;1H NMR(500MHz,CDCl3)δ:9.5(1H,s),7.21(1H,br s),6.17(1H,br s),5.05(1H,br s),4.08(1H,dd,J=11.7,2.7Hz),3.84(1H,d,J=11.7Hz),3.52(3H,s),2.73(1H,dd,J=16.3,6.0Hz),2.63(1H,m),2.07‐2.36(4H,m),1.46‐1.83(8H,m),1.22‐1.39(2H,m),0.95(3H,d,J=7.1Hz).13C NMR(125MHz,CDCl3)δ:205.1(d),148.7(s),140.7(d),122.1(s),109.6(d),100.5(d),67.2(t),57.2(q),51.7(s),45.9(d),41.3(d),38.3(t),36.4(d),35.2(s),31.5(d),29.9(t),27.5(t),22.5(t),22.3(t),20.7(t),17.1(q).正离子ESIMS m/z367[M+Na]+;HRESIMS m/z367.1886[M+Na]+(calcd for C21H28O4Na,367.1885).
实施例12:
化合物13的制备
化合物P1(20mg)溶于6mL四氢呋喃溶剂中,加入KOH(10mg)于75℃下加热反应3h。冷却至室温,向反应液中加入1mL1N盐酸溶液搅拌5分钟,用乙酸乙酯萃取,萃取液用饱和碳酸氢钠溶液洗涤3次,无水硫酸钠干燥,减压浓缩。浓缩后的样品继续溶于5mL二氯甲烷溶剂中,加入PDC(30mg)于室温反应12h。加入适量饱和碳酸氢钠溶液,用乙酸乙酯萃取3次,水硫酸钠干燥,减压浓缩得淡黄色固体。经硅胶柱层析(石油醚:乙酸乙酯=8:2)得到化合物13(12mg,产率73%)。
化合物13:白色粉末;1H NMR(500MHz,CDCl3)δ:7.22(1H,br s),6.18(1H,br s),4.58(1H,dd,J=11.9,2.1Hz),4.23,(1H,d,J=11.9Hz),2.58‐2.81(2H,m),1.99‐2.35(4H,m),1.57‐1.91(7H,m),1.17‐1.54(4H,m),1.00(3H,d,J=7.1Hz).13C NMR(125MHz,CDCl3)δ:173.8(s),148.1(s),140.9(d),122.6(s),109.6(d),73.8(t),43.8(d),43.7(d),40.5(d),38.5(t),36.5(d),31.4(d),30.3(t),29.4(t),22.4(t),20.4(t),17.4(q).正离子ESIMS m/z323[M+Na]+.
实施例13:
化合物1‐13对糖异生的抑制作用:
(1)实验方法:
a、大鼠禁食24h后经500mg/kg的水合氯醛腹腔注射麻醉后,全身酒精消毒,迅速开腹,门静脉插管固定,用的前灌流液以10~15ml/min灌流,同时剪断下腔静脉,约10mins。
b、换含胶原酶的后灌流溶液以5~7ml/min灌流,灌流至肝包膜下组织呈龟背状裂隙或门静脉破裂时中止,时间约15~20min。
c、灌流结束后,分离完整肝脏,经MEM培养液清洗后置于含相同培液的培养皿中,用镊子撕去肝包膜,用滴管轻轻吹打分散后,100目筛网过滤,将滤液吸到50ml离心管中,离心500rpm,3min,弃上清。
d、Percoll分离细胞,离心500rpm,5min。吸去上清液和死细胞。加入10%FBSMEM培养液(含10nM insulin和10nM DEX)悬浮细胞,经0.4%台盼蓝进行细胞活性检测并计数,按1.3×105个/well细胞接种于铺有明胶的48‐well培养板中,置于5%CO2培养箱中培养。
e、大鼠原代肝细胞培养4h,贴壁后,换入含有不同浓度的化合物、0.1%DMSO(溶剂对照)或500μM Metformin(阳性对照)的无糖DMEM培养液,预处理1.5h后,换入含有活不含有糖异生底物(20mM乳酸钠,2mM丙酮酸钠)及不同浓度的化合物、0.1%DMSO(溶剂对照)或500μM Metformin(阳性对照)的无糖DMEM溶液。孵育4h后,收集培液,检测器葡萄糖浓度。同时,用PBS洗细胞3遍后加入0.5M NaOH裂解细胞,测定蛋白浓度并经蛋白浓度校正葡萄糖读数后求得单位细胞数所产生葡萄糖的量。
(2)实验结果:
实验结果表明化合物1‐13对肝糖异生具有显著抑制作用,具有治疗2型糖尿病及其他代谢性疾病的潜力。
表1.本发明化合物对肝细胞糖异生的影响
a阳性对照二甲双胍500μM时糖异生值为0.61
实施例14:
片剂的制备:
按实施例1‐12的方法先制得本发明的上述化合物,按其与赋形剂重量比为1:5‐1:10的比例加入赋形剂,制粒压片。
实施例15:
口服液制剂的制备:
按实施例1‐12的方法先制得本发明的上述化合物,以及利用有机酸(酒石酸,柠檬酸,甲酸,乙二酸等)或无机酸(盐酸,硫酸,磷酸等)制成的盐,按常规口服液制法制成口服液。
实施例16:
胶囊剂、颗粒剂、或冲剂的制备:
按实施例1‐12的方法先制得本发明的上述化合物,按其与赋形剂重量比为5:1的比例加入赋形剂,制成胶囊或颗粒剂或冲剂。
- 还没有人留言评论。精彩留言会获得点赞!
- 基于四氢呋喃修饰核苷酸及其在dna测序中的用图
- 用钌/双齿配体络合物进行的醛的选择性氢化的制作方法
- 二氢杨梅素-钙络合物的制备方法
- 一种促进Ru(II)金属络合物细胞核摄取的方法
- 一种6-(5-氯-2-吡啶基)-5-羟基-7-氧代-6,7-二氢-5H-吡咯并[3,4b]吡嗪的制备方法
- 一种合成雷美替胺的关键中间体2-(2,6,7,8-四氢-1H-茚并[5,4-b]呋喃-8-基)丙烯酸乙 ...的制作方法
- 一种用质子响应型铱配合物催化氨硼烷水解制氢的方法
- 第5族金属氧代-烷氧代络合物、其制造方法以及第5族金属氧化物膜的制作方法
- 常温直接制备三氯化镓-四氢呋喃溶液的装置的制造方法
- 生产用于选择性提取铟的络合物形成吸附剂的方法
- 瑞香烷型二萜化合物pimelotide C的新用途的制作方法与工艺
- 大别山五针松的树皮提取物及其制备方法和在制药中的用途与流程
- 雷公藤二萜合酶TwCPS1在制备松香烷型二萜化合物中的应用的制造方法与工艺
- 雷公藤焦磷酸合酶TwCPS4及其制备松香烷型二萜化合物的应用的制造方法与工艺
- 新的抗炎松香烷型二萜糖苷tripterycosideC的制造方法与工艺
- 新的抗炎松香烷型二萜糖苷tripterycosideB的制造方法与工艺
- 新的抗炎松香烷型二萜糖苷tripterycosideA的制造方法与工艺
- 松香烷型二萜糖苷在制备抗炎药物中的应用的制造方法与工艺
- 羟基化续随子二萜烷型衍生物的转化方法及其在制备抗肿瘤药物中的用途与流程
- 一种新的半日花烷型二萜化合物及其制备方法和应用、药物组合物及其应用与制造工艺