一种纳米复合物及其制备方法与应用与流程
本发明属于高分子化学与生物医学工程领域,尤其涉及一种纳米复合物及其制备方法与应用。
背景技术:
阳离子聚合物载体是非病毒载体中的重要一类,此类材料能够通过静电相互作用和带负电荷的核酸分子相结合,确保核酸分子在输送过程中的完整性。相对于病毒载体,非病毒载体的发展面对的一个主要挑战就是:如何提高转染效率。
溶酶体逃逸是基于阳离子载体的核酸输送效率提高的一个关键点。一般情况下,如果细胞内吞的纳米颗粒物转运到溶酶体中,将面临被溶酶体水解酶降解的危险。因此如果polyplexes细胞内化转运到溶酶体中时,必须设法突破溶酶体膜的屏障,尽早地实现溶酶体逃逸。当前阳离子聚合物核酸载体材料聚乙烯胺(pei)被公认为基因转染的“黄金标准”,主要是由于pei具有较强的“质子海绵”效应,能够有效的实现溶酶体逃逸,获得较高的转染水平。在生理ph7.4条件下,pei只有15~20%的氨基可以被质子化;但在溶酶体通路中,随着ph的降低,pei上氨基的质子化程度显著提高,导致纳米复合物的表面正电荷密度上升,对溶酶体膜的扰动作用增强。同时,由于质子化过程引起大量质子和氯离子的流入,提高了溶酶体内的渗透压,进一步降低了溶酶体膜的稳定性,有助于纳米复合物实现溶酶体逃逸和转运细胞的表达效率。
除了质子海绵效应外,光化学内化(photochemicalinternalization,pci)技术也是一个辅助大分子及纳米粒子实现溶酶体逃逸的有效策略。pci技术基于光动力学治疗(photodynamictherapy,pdt)的原理,通过使用光、氧气和光敏剂相互作用促进大分子及纳米粒子的溶酶体逃逸。通常,pci策略是通过纳米复合物体系中引入卟啉、酞菁等具有强光诱导单线态氧产生能力的光敏剂,从而增强溶酶体膜的通透性,提高纳米粒子的细胞质释放率。
共轭聚电解质材料(conjugatedpolyelectrolyte,cpe)是一类具有由不饱和键和单键交替排列而成的主链和离子化侧链的聚合物,同时具备独特的光学性质及良好的亲水性,在生物医学领域受到研究者们的青睐。例如,阳离子聚噻吩(cationicpolythiophene,cpt)是一类易于制备、生物兼容性良好、光学性质优异的共轭聚电解质材料,在细胞分化、生物传感器、荧光探针、荧光成像、仿生人造器官等生物领域具有广泛的应用价值。类似的,阳离子聚苯撑乙炔(cationicpoly(phenyleneethynylene),cppe)也被应用于生物传感、荧光成像、抗菌等生物医学领域。cpe一般具有对氧分子的光敏化能力。
通常,阳离子聚合物载体材料往往需要相对较高的核酸用量以及较多的聚合物过量使用(96孔板中,剂量一般为5μg/ml或以上,n/p一般为10或以上;n/p越高,意味着载体材料用量越大),既不经济,也不安全。众所周知,使用过多的载体材料是引起细胞毒性最主要的原因。但是在阳离子聚合物载体体系中存在的普遍问题是,一旦降低聚合物和核酸分子的用量,就会导致核酸输送效率严重偏低的不利结果。
因此,在聚合物载体材料和核酸分子用量双降的前提下,采取新的聚合物载体体系构建策略,开发核酸输送的有效增强方法,将使聚合物载体材料在生物医学应用领域具有更广泛的应用价值。
技术实现要素:
发明目的:本发明的第一目的是提供一种能够在低质粒和聚合物用量的条件下有效增强核酸转运效率的纳米复合物;
本发明的第二目的是提供该复合物的制备方法;
本发明的第三目的是提供该复合物的应用。
技术方案:本发明的纳米复合物,包括:阳离子聚合物载体、核酸分子及共轭聚电解质;其中,共轭聚电解质及阳离子聚合物载体的正电荷总和与核酸分子的负电荷之比为1~20:1,共轭聚电解质与阳离子聚合物载体的正电荷之比为0.01~10%。
本发明添加微量共轭聚电解质,通过共轭聚电解质材料的光敏化产生ros的作用,增强溶酶体膜通透性的改变、辅助纳米复合物的溶酶体逃逸。
进一步说,所述阳离子聚合物载体包括聚氨基酸衍生物、聚糖衍生物或聚酰胺衍生物。优选的,聚氨基酸衍生物为可降解阳离子星形聚天冬酰胺载体材料,其由聚天冬酰胺主链和二乙烯三胺侧链构成,结构式如下所示:
再进一步说,所述共轭聚电解质包括阳离子聚噻吩、阳离子聚苯撑乙炔、阳离子聚芴、阳离子聚苯撑乙烯或阳离子聚电解质共聚物。优选的,阳离子聚噻吩为阳离子线形聚噻吩或阳离子支化聚噻吩。其中,阳离子线形聚噻吩的结构式为:
r为
阳离子支化聚噻吩的结构式为:
其中,m/n=1/10~1/100,n=10~250,
r为
优选的,阳离子聚苯撑乙炔的结构式为:
其中,n=10~250。
本发明制备纳米复合材料的方法,包括如下步骤:将阳离子聚合物载体与核酸分子混合,加入共轭聚电解质,静置10~60min,得到多组份纳米复合物。
本发明所述的纳米复合物应用于增强核酸的输送。
有益效果:与现有技术相比,本发明的显著优点为:通过在聚合物载体及核酸分子体系基础上,引入微量共轭聚电解质材料,形成该纳米复合物,采用非共价组装途径对纳米复合物表面进行功能化改造,不仅稳定性强,且应用于核酸的输送中,在光照或非光照条件下均能够增强溶酶体膜的渗透性,帮助纳米复合物实现溶酶体逃逸,不会对细胞造成显著损伤,增强基因输送及核酸转运效率,满足基因输送安全高效的要求;同时,其制备方法设计合理,制备过程简单,大幅减少载体材料的使用量,具有广阔的应用前景。
附图说明
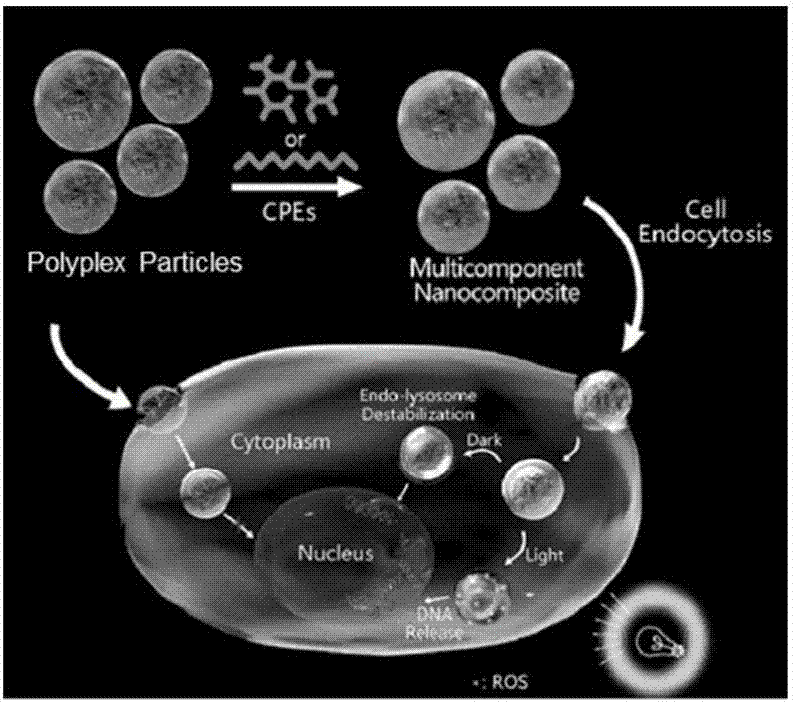
图1为多组份纳米复合物体系修饰增强核酸输送的示意图;
图2a为阳离子线性聚噻吩的制备工艺流程图;
图2b为阳离子支化聚噻吩的制备工艺流程图;
图3a为lptm和bptm在cdcl3中的1hnmr表征图;
图3b为lpt和bpt在cdcl3中的1hnmr表征图;
图3c为脂溶性线性/支化聚噻吩的gpc表征,thf为流动相图;
图3d为脂溶性线性/支化聚噻吩由gpc测试结果计算得出的分子量和pdi图;
图4为阳离子聚噻吩构建的多组份纳米复合物的尺寸和表面电势图;
图5为阳离子聚噻吩构建的多组份纳米复合物的eb竞争实验图;
图6为阳离子线性/支化聚噻吩构建的多组份纳米复合物对hepg2细胞作用24h的细胞毒性图;
图7a为无光照条件下,阳离子星形聚天冬酰胺载体材料(sp)在hela细胞内引起的光诱导ros检测结果图;
图7b为以dcfh-da为ros荧光成像探针,光照条件下,阳离子星形聚天冬酰胺载体材料(sp)在hela细胞内引起的光诱导ros检测结果图;
图8a为无光照条件下,聚乙烯胺载体材料(pei)在hela细胞内引起的光诱导ros检测结果图;
图8b为以dcfh-da为ros荧光成像探针,光照条件下,聚乙烯胺载体材料(pei)在hela细胞内引起的光诱导ros检测结果图;
图9a为无光照条件下,5%的阳离子支化聚噻吩(cbptm-1)构建的多组份纳米复合物在hela细胞内引起的光诱导ros检测结果图;
图9b为以dcfh-da为ros荧光成像探针,光照条件下,5%的阳离子支化聚噻吩(cbptm-1)构建的多组份纳米复合物在hela细胞内引起的光诱导ros检测结果图;
图10a为无光照条件下,2.5%的阳离子支化聚噻吩(cbptm-1)构建的多组份纳米复合物在hela细胞内引起的光诱导ros检测结果图;
图10b为以dcfh-da为ros荧光成像探针,光照条件下,2.5%的阳离子支化聚噻吩(cbptm-1)构建的多组份纳米复合物在hela细胞内引起的光诱导ros检测结果图;
图11a为无光照条件下,5%的阳离子线性聚噻吩(clptm)构建的多组份纳米复合物在hela细胞内引起的光诱导ros检测结果图;
图11b为以dcfh-da为ros荧光成像探针,光照条件下,5%的阳离子线性聚噻吩(clptm)构建的多组份纳米复合物在hela细胞内引起的光诱导ros检测结果图;
图12a为无光照条件下,2.5%的阳离子线性聚噻吩(clptm)构建的多组份纳米复合物在hela细胞内引起的光诱导ros检测结果图;
图12b为以dcfh-da为ros荧光成像探针,光照条件下,2.5%的阳离子线性聚噻吩(clptm)构建的多组份纳米复合物在hela细胞内引起的光诱导ros检测结果图;
图13a为无光照条件下,5%的阳离子线性聚噻吩(clpt-1)构建的多组份纳米复合物在hela细胞内引起的光诱导ros检测结果图;
图13b为以dcfh-da为ros荧光成像探针,光照条件下,5%的阳离子线性聚噻吩(clpt-1)构建的多组份纳米复合物在hela细胞内引起的光诱导ros检测结果图;
图14a为无光照条件下,2.5%的阳离子线性聚噻吩(clpt-1)构建的多组份纳米复合物在hela细胞内引起的光诱导ros检测结果图;
图14b为以dcfh-da为ros荧光成像探针,光照条件下,2.5%的阳离子线性聚噻吩(clpt-1)构建的多组份纳米复合物在hela细胞内引起的光诱导ros检测结果图;
图15a为无光照条件下,5%的阳离子支化聚噻吩(cbpt-1)构建的多组份纳米复合物在hela细胞内引起的光诱导ros检测结果图;
图15b为以dcfh-da为ros荧光成像探针,光照条件下,5%的阳离子支化聚噻吩(cbpt-1)构建的多组份纳米复合物在hela细胞内引起的光诱导ros检测结果图;
图16a为无光照条件下,2.5%的阳离子支化聚噻吩(cbpt-1)构建的多组份纳米复合物在hela细胞内引起的光诱导ros检测结果图;
图16b为以dcfh-da为ros荧光成像探针,光照条件下,2.5%的阳离子支化聚噻吩(cbpt-1)构建的多组份纳米复合物在hela细胞内引起的光诱导ros检测结果图;
图17a为无光照条件下,hela在无血清培养基中与聚乙烯胺载体材料(pei)共育5h后,在绿色通道的溶酶体通透性检测结果图;
图17b为无光照条件下,hela在无血清培养基中与聚乙烯胺载体材料(pei)共育5h后,在红色通道的溶酶体通透性检测结果图;
图17c为无光照条件下,hela在无血清培养基中与聚乙烯胺载体材料(pei)共育5h后,在合并通道的溶酶体通透性检测结果图;
图17d为光照条件下,hela在无血清培养基中与聚乙烯胺载体材料(pei)共育5h后,在绿色通道的溶酶体通透性检测结果图;
图17e为光照条件下,hela在无血清培养基中与聚乙烯胺载体材料(pei)共育5h后,在红色通道的溶酶体通透性检测结果图;
图17f为光照条件下,hela在无血清培养基中与聚乙烯胺载体材料(pei)共育5h后,在合并通道的溶酶体通透性检测结果图;
图18a为无光照条件下,hela在无血清培养基中与阳离子星形聚天冬酰胺载体材料(sp)共育5h后,在绿色通道的溶酶体通透性检测结果图;
图18b为无光照条件下,hela在无血清培养基中与阳离子星形聚天冬酰胺载体材料(sp)共育5h后,在红色通道的溶酶体通透性检测结果图;
图18c为无光照条件下,hela在无血清培养基中与阳离子星形聚天冬酰胺载体材料(sp)共育5h后,在合并通道的溶酶体通透性检测结果图;
图18d为光照条件下,hela在无血清培养基中与阳离子星形聚天冬酰胺载体材料(sp)共育5h后,在绿色通道的溶酶体通透性检测结果图;
图18e为光照条件下,hela在无血清培养基中与阳离子星形聚天冬酰胺载体材料(sp)共育5h后,在红色通道的溶酶体通透性检测结果图;
图18f为光照条件下,hela在无血清培养基中与阳离子星形聚天冬酰胺载体材料(sp)共育5h后,在合并通道的溶酶体通透性检测结果图;
图19a为无光照条件下,hela在无血清培养基中与阳离子线性聚噻吩(clpt-2)共育5h后,在绿色通道的溶酶体通透性检测结果图;
图19b为无光照条件下,hela在无血清培养基中与阳离子线性聚噻吩(clpt-2)共育5h后,在红色通道的溶酶体通透性检测结果图;
图19c为无光照条件下,hela在无血清培养基中与阳离子线性聚噻吩(clpt-2)共育5h后,在合并通道的溶酶体通透性检测结果图;
图19d为光照条件下,hela在无血清培养基中与阳离子线性聚噻吩(clpt-2)共育5h后,在绿色通道的溶酶体通透性检测结果图;
图19e为光照条件下,hela在无血清培养基中与阳离子线性聚噻吩(clpt-2)共育5h后,在红色通道的溶酶体通透性检测结果图;
图19f为无光照条件下,hela在无血清培养基中与阳离子线性聚噻吩(clpt-2)共育5h后,在合并通道的溶酶体通透性检测结果图;
图20为阳离子线性/支化聚噻吩与sp构建的多组份纳米复合物对hela细胞转染表达luciferase的质粒pluci40h的转染情况图;
图21a为bpei在无光照条件下对hela细胞转染表达绿色荧光蛋白的质粒pgfp40h的转染情况图;
图21b为bpei在光照条件下对hela细胞转染表达绿色荧光蛋白的质粒pgfp40h的转染情况图;
图22a为sp在无光照条件下对hela细胞转染表达绿色荧光蛋白的质粒pgfp40h的转染情况图;
图22b为sp在光照条件下对hela细胞转染表达绿色荧光蛋白的质粒pgfp40h的转染情况图;
图23a为0.1%的阳离子聚苯撑乙炔(pim-1)构建的多组份纳米复合物在无光照条件下对hela细胞转染表达绿色荧光蛋白的质粒pgfp40h的转染情况图;
图23b为0.1%的阳离子聚苯撑乙炔(pim-1)构建的多组份纳米复合物在光照条件下对hela细胞转染表达绿色荧光蛋白的质粒pgfp40h的转染情况图;
图24a为0.25%的阳离子聚苯撑乙炔(pim-1)构建的多组份纳米复合物在无光照条件下对hela细胞转染表达绿色荧光蛋白的质粒pgfp40h的转染情况图;
图24b为0.25%的阳离子聚苯撑乙炔(pim-1)构建的多组份纳米复合物在光照条件下对hela细胞转染表达绿色荧光蛋白的质粒pgfp40h的转染情况图;
图25a为0.5%的阳离子聚苯撑乙炔(pim-1)构建的多组份纳米复合物在无光照条件下对hela细胞转染表达绿色荧光蛋白的质粒pgfp40h的转染情况图;
图25b为0.5%的阳离子聚苯撑乙炔(pim-1)构建的多组份纳米复合物在光照条件下对hela细胞转染表达绿色荧光蛋白的质粒pgfp40h的转染情况图;
图26a为1%的阳离子聚苯撑乙炔(pim-1)构建的多组份纳米复合物在无光照条件下对hela细胞转染表达绿色荧光蛋白的质粒pgfp40h的转染情况图;
图26b为1%的阳离子聚苯撑乙炔(pim-1)构建的多组份纳米复合物在光照条件下对hela细胞转染表达绿色荧光蛋白的质粒pgfp40h的转染情况图;
图27为无光照条件下,阳离子聚苯撑乙炔(pim-1、pim-2)与sp构建的多组份纳米复合物对hela细胞转染表达luciferase的质粒pluci40h的转染情况图,bpei和sp的转染情况作为对照。
具体实施方式
下面通过实施例及附图对本发明的技术方案进行详细说明。
本发明的纳米复合物,包括:阳离子聚合物载体、核酸分子及共轭聚电解质;其中,共轭聚电解质及阳离子聚合物载体的正电荷总和与核酸分子的负电荷之比为1~20:1,共轭聚电解质与阳离子聚合物载体的正电荷之比为0.01~10%。
其中,阳离子聚合物载体包括聚氨基酸衍生物、聚糖衍生物或聚酰胺衍生物等。具体而言,阳离子聚合物载体包括骨架及侧链,其中,阳离子聚合物载体的骨架包括聚氨基酸、聚丙烯酰胺、聚甲基丙烯酰胺、聚丙烯酸酯、聚甲基丙烯酸酯、聚降冰片烯、聚乙烯胺、聚糖、聚酰胺、或基于上述骨架结构的共聚物等;阳离子聚合物侧链基团包括烷基胺、烷基季铵盐、烷基咪唑等,其中烷基可以是c1~c18链,烷基链上含有0~6个杂原子,聚合物分子量为2000~150000da。
共轭聚电解质包括阳离子聚噻吩、阳离子聚苯撑乙炔、阳离子聚芴、阳离子聚苯撑乙烯或阳离子聚电解质共聚物。具体而言,共轭聚电解质包括骨架及侧链,其中,共轭聚电解质骨架包括聚噻吩、聚苯撑乙炔、聚芴、聚苯撑乙烯、聚丁二炔、或基于上述骨架结构的共聚物等;共轭聚电解质侧链基团包括烷基胺、烷基季铵盐、烷基咪唑等,其中烷基可以是c1~c18链,链上含有0~6个杂原子。聚合物分子量为1000~50000da。优选的,阳离子聚噻吩可包括线形阳离子聚噻吩或阳离子支化聚噻吩;阳离子聚苯撑乙炔可包括线形阳离子聚苯撑乙炔或阳离子支化聚苯撑乙炔;阳离子聚芴可为线形阳离子聚芴;阳离子聚苯撑乙烯可为线形阳离子聚苯撑乙烯;阳离子聚电解质共聚物可包括阳离子聚(苯撑乙炔-co-苯)、阳离子聚(芴-co-苯)等。
进一步说,本发明阳离子线性聚噻吩由如下步骤制备而得:
(1)分别催化氧化3-噻吩乙酸乙酯或者3-噻吩丙二酸二乙酯单体聚合,随后清洗使铁离子完全除去,得到两类脂溶性线性聚噻吩;
(2)将上述两类脂溶性线性聚噻吩分别经分离纯化、胺解、质子化及透析后,即得到阳离子线性聚噻吩。
本发明阳离子支化聚噻吩由如下步骤制备而得:
(1)按摩尔比0.5~1:1将2-噻吩乙酸与n-羟基琥珀酰亚胺反应生成2-噻吩乙酸活性酯;
(2)将2-噻吩乙酸活性酯与三(2-氨乙基)胺及三乙胺在ch2cl2中反应,制得如下结构式的t3,其中,2-噻吩乙酸活性酯、三(2-氨乙基)胺、三乙胺的摩尔比为0.1~1:0.03~0.3:1,
(3)分别催化氧化噻吩乙酸乙酯与t3或者噻吩丙二酸二乙酯与t3聚合,随后清洗,使铁离子完全除去得到两类脂溶性支化聚噻吩,其中,噻吩乙酸乙酯与t3的摩尔比为10~100:1,噻吩丙二酸二乙酯与t3的摩尔比为10~100:1;
(4)将上述两类脂溶性支化聚噻吩分别经分离纯化、胺解、质子化及透析后,即得到阳离子支化聚噻吩。
本发明阳离子聚苯撑乙炔cppe-det由如下步骤制备而得:
(1)将2,5-二碘-1,4-二苯乙酸溶于无水乙醇中,加入二氯亚砜,制得2,5-二碘-1,4-二苯乙酸乙酯,其中,2,5-二碘-1,4-二苯乙酸与二氯亚砜的摩尔比为10~100:1;
(2)将2,5-二碘-1,4-二苯乙酸乙酯溶于无水四氢呋喃和三乙胺中,分别加入pd(pph3)4、cui及三甲基硅烷乙炔,随后加入四丁基氟化铵,沉淀制得ppe-et,其中,2,5-二碘-1,4-二苯乙酸乙酯、pd(pph3)4、cui及三甲基硅烷乙炔的摩尔比为0.25~0.5:0.001~0.01:0.001~0.01:1;
(3)将上述制得的ppe-et溶于nmp,加入二乙基三胺的nmp溶液,透析后制得cppe-det。
本发明阳离子聚苯撑乙烯由如下步骤制备而得:
(1)将2,5-二碘-1,4-二苯乙酸溶于无水乙醇,加入二氯亚砜,制得2,5-二碘-1,4-二苯乙酸乙酯,其中,2,5-二碘-1,4-二苯乙酸与二氯亚砜的摩尔比为10~100:1;
(2)将2,5-二碘-1,4-二苯乙酸乙酯、1,4-苯二乙烯溶于无水二氧六环,加入三乙胺和pd/c,反应制得ppv-et,其中,2,5-二碘-1,4-二苯乙酸乙酯、1,4-苯二乙烯的摩尔比为0.8~1.25:1;
(3)将上述ppv-et溶于nmp,加入二乙基三胺的nmp溶液,即制得cppv-det。
本发明阳离子共聚物聚(芴-co-苯)由如下步骤制备而得:
将fl-et、1,4-二苯硼酸溶于甲苯和碳酸钾水溶液中,随后加入四(三苯基膦)钯,反应后制得pfp-et,将该pfp-et溶于干燥的nmp,加入r-nh2的nmp溶液,反应后制得cpfp-r,其中,fl-et、1,4-二苯硼酸、四(三苯基膦)钯的摩尔比为10~25:10~25:1。
下述实施例中,如无特殊说明,均为常规方法,所用的实验材料,如无特殊说明,均自常规生化试剂商店购买得到的。下述实施例中所述x%的共轭聚电解质用量,如无特殊说明,均为摩尔量的百分含量。以下实施例中的定量实验,均设置三次重复试验,结果取平均值。
实施例1阳离子聚噻吩的制备
1、阳离子线性聚噻吩
本发明的阳离子线性聚噻吩的结构式为:
制备方法包括如下步骤:
(1)脂溶性线性聚噻吩(lpt或lptm)的合成
氮气保护下,将无水三氯化铁(1.05g,6.5mmol)溶于无水硝基甲烷(25ml),另在氮气保护下,将3-噻吩乙酸乙酯(0.50g,3.34mmol)或者3-噻吩丙二酸二乙酯(0.80g,3.31mmol)溶解于无水chcl3(25ml);冰浴下,将fecl3的硝基甲烷溶液逐滴加入反应体系,体系加热至35℃,避光搅拌反应2h,停止反应后,体系先用0.01m盐酸洗一遍,再用含有三乙醇胺(5%,w/v)和柠檬酸钠(36.7g/l)的混合溶液洗三次,最后用氟化铵(10%,w/v)洗一次,使铁离子完全除去;有机相用无水硫酸钠干燥过夜后蒸除溶剂,得到的固体用少量氯仿溶解,再用乙醚重沉淀,得到脂溶性线性聚噻吩的混合物,该混合物进一步分别用尺寸排阻色谱法(sec)分离,得到脂溶性线性聚噻吩(lpt1、lpt2、lptm)。
(2)阳离子线性聚噻吩(clpt或clptm)的合成
称取上述脂溶性线性聚噻吩(0.50g)溶于nmp(10ml)中,冰箱内放置6h,使其充分溶解;氩气保护下,另取二乙烯三胺det(50eq.)溶于n-甲基吡咯烷酮,即nmp(8ml)中,并用冰盐浴冷却,随后,搅拌下,将已置换氩气的脂溶性线性聚噻吩的nmp溶液逐滴加入至det的nmp溶液中,反应4h,反应停止后,将体系慢慢逐滴加入预冷的ph=1的盐酸中,冰盐浴下大力搅拌;将盐酸质子化后得到澄清透明的液体转入透析袋(根据gpc测试结果确定使用何种截留分子量的透析袋)中,先用ph=2的盐酸溶液透析1d,再用ph=4的盐酸溶液透析1d,最后用中性去离子水透析2d,透析完成后,取出透析袋中液体,用0.22μm滤器过滤后,冻干得到阳离子线性聚噻吩(clptm、clpt-1、clpt-2)。
上述反应的流程如图2a所示。
2、阳离子支化聚噻吩
本发明的阳离子支化聚噻吩的结构式为:
制备方法包括如下步骤:
(1)化合物t3的合成
将2-噻吩乙酸(0.30g,2.1mmol)溶于二氯甲烷(20ml),加入1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(0.48g,2.52mmol)及n-羟基琥珀酰亚胺(0.29g,2.52mmol),室温下反应过夜;水洗、无水硫酸钠干燥后,旋蒸除去溶剂,将上述2-噻吩乙酸活性酯溶于二氯甲烷(8ml),加入三(2-氨乙基)胺(0.08g,0.55mmol)和三乙胺(0.067g,0.66mmol)并搅拌反应24h,反应结束后,反应体系用饱和氯化铵水溶液洗两次,饱和食盐水洗一次,无水硫酸钠干燥过夜,产物浓缩后用硅胶柱分离纯化,得到白色固体t3(0.18g);将t3进行性能检测,可得到如下所述的产物表征:核磁氢谱(400mhz,cdcl3):δ(ppm)7.19(3h),6.96-6.90(m,6h),6.70(brs,3h),3.75(s,6h),3.19(q,6h),2.47(t,6h).核磁碳谱(75mhz,dmso-d6):δ(ppm)169.7,138.1,127.0,126.4,125.2,53.7,37.6,37.0.高分辨质谱:m/zc24h31n4o3s3([m+h]+)计算值519.1558,实测值519.1557。上述表征数据表明t3结构无误。
(2)脂溶性支化聚噻吩(bpt或bptm)的合成。
氮气保护下,将无水三氯化铁(1.05g,6.5mmol)溶解于无水硝基甲烷(25ml);另在氮气保护下,将3-噻吩乙酸乙酯(0.50g,3.3mmol)或者3-噻吩丙二酸二乙酯(0.80g,3.3mmol)和t3(0.07g,0.14mmol)溶解于无水chcl3(25ml),冰浴下,将fecl3的硝基甲烷溶液逐滴加入反应体系,体系升温至35℃,避光搅拌反应2h;停止反应后,体系先用0.01m盐酸洗一遍,再用含有三乙醇胺(5%,w/v)和柠檬酸钠(36.7g/l)的混合溶液洗三次,最后用氟化铵(10%,w/v)洗一次,使铁离子完全除去;有机相用无水硫酸钠干燥过夜后蒸除溶剂,得到的固体用少量氯仿溶解,再用乙醚重沉淀,得到脂溶性线性聚噻吩的混合物,该混合物进一步用尺寸排阻色谱法分离,收集各部分产物并分别浓缩干燥,即得到脂溶性支化聚噻吩bpt-1、bpt-2、bpt-3、bptm-1、bptm-2。
上述lpt1、lpt2、lptm的1hnmr和gpc的表征结果,bpt或bptm的1hnmr和gpc的表征结果如图3a至3d所示。根据lpt1、lpt2、lptm的1hnmr和gpc的表征结果可知,低场、中高场以及高场的共振峰对应核磁谱图中噻吩单元的氢,以聚苯乙烯为参照物的gpc分析显示mw分子量分别为33、9、4kda,说明为聚噻吩结构;根据bpt或bptm的1hnmr和gpc的表征结果可知,在核磁氢谱方面,bpt和bptm除了具有噻吩单元的共振峰,还具有支化单元t3的共振峰,在gpc方面,bpt和bptm都出现在比单体更短的保留时间,上述结果表明bpt和bptm为聚合产物,且t3结构被成功整合到聚噻吩中。
(3)阳离子支化聚噻吩(cbpt或cbptm)的合成
称取上述脂溶性支化聚噻吩(0.50g)溶于nmp(10ml)中,冰箱内放置6h,使其充分溶解;氩气保护下,另取det(50eq.)溶于nmp(8ml)中并用冰盐浴冷却,随后,搅拌下,将已置换氩气的脂溶性支化聚噻吩的nmp溶液逐滴加入至det的nmp溶液中,反应4h;反应停止后,将体系慢慢逐滴加入预冷的ph=1的盐酸中,冰盐浴下大力搅拌。将盐酸质子化后得到澄清透明的液体转入透析袋(根据gpc测试结果确定使用何种截留分子量的透析袋)中,先用ph=2的盐酸溶液透析1d,再用ph=4的盐酸溶液透析1d,最后用中性去离子水透析2d,透析完成后,取出透析袋中液体,用0.22μm滤器过滤后,冻干得到阳离子支化聚噻吩(cbptm-1、cbptm-2、cbpt-1、cbpt-2、cbpt-3)。其制备工艺流程图如图2b所示。
在制备阳离子支化聚噻吩时,步骤(1)中,将2-噻吩乙酸与n-羟基琥珀酰亚胺的摩尔比替换成0.5:1或1:1,其中,2-噻吩乙酸活性酯与三(2-氨乙基)胺及三乙胺的摩尔比为0.1:0.03:1、0.5:0.1:1或1:0.3:1,均可制得该结构的阳离子支化聚噻吩;步骤(2)中,将噻吩乙酸乙酯与t3,或噻吩丙二酸二乙酯与t3的摩尔比替换为10:1、50:1、100:1,均可制得该结构的阳离子支化聚噻吩。
实施例2阳离子线性/支化聚噻吩构建的多组份纳米复合物
原料组成:该复合物包括可降解阳离子星形聚天冬酰胺载体、dna及阳离子线性/支化聚噻吩;其中,阳离子线性/支化聚噻吩及可降解阳离子星形聚天冬酰胺载体的正电荷总和与dna的负电荷之比为2.5:1,阳离子线性/支化聚噻吩与可降解阳离子星形聚天冬酰胺载体的正电荷之比为2.5%或5%。
制备方法:在聚合物材料所含的胺基(包括1~4级胺)数目与核酸分子所含磷酸酯键数目之比为5的条件下(即n/p=5,由于只有一半胺基在中性条件下发生了质子化,此时正负电荷比为2.5:1),首先将可降解阳离子星形聚天冬酰胺载体(sp)材料和质粒dna用超纯水稀释到相同的体积后,将质粒溶液缓慢加入到sp材料中,完全加完后静置15min,使材料与质粒自组装形成稳定的纳米复合物。随后,向纳米复合物体系中缓慢加入等体积的阳离子线性/支化聚噻吩,完全加完后再静置15min,使阳离子线性/支化聚噻吩结合在纳米复合物表面,组装形成稳定的多组份纳米复合物。
引入2.5%、5%阳离子线性/支化聚噻吩构建的多组份纳米复合物体系,测定其水合动力学直径及表面电势,结果如图4所示,加入少量的阳离子线性/支化聚噻吩后,纳米复合物的尺寸变小,表面电势提高。
如图1所示,核酸分子被稳定包装于内核处的纳米复合材料被细胞摄取后,其表面的共轭聚电解质在非光照下扰动溶酶体膜,或者在光照下通过光化学内化效应进一步增加溶酶体膜的通透性,从而显著提高核酸颗粒溶酶体逃逸的成功率以及最终的转染效率。
性能检测1阳离子线性/支化聚噻吩构建的多组份纳米复合物的稳定性检测
溴化乙锭(ethidiumbromide,eb)竞争实验用于检测阳离子线性/支化聚噻吩构建的多组份纳米复合物的稳定性。实验中eb终浓度为5μg/ml,质粒dna终浓度为1.6μg/ml,去离子为溶剂。以n/p=0.25为变化当量,逐步加入材料(加入材料溶液的体积尽量少,以免引起质粒浓度较大的变化),混匀后静置5min测定eb的荧光发射光谱强度变化,溶液随着材料加入,体系相对荧光强度(relativefluorescenceintensity,rfi)可以按如下公式计算得出:
rfi=(fx-f0)/(fmax-f0)
其中,f0表示仅有eb时,体系荧光强度;fmax表示eb中加入质粒dna,稳定5min后的荧光强度;fx表示eb与质粒dna的溶液中加入载体材料,稳定5min后荧光强度。为了避免阳离子噻吩材料的干扰,选用激发光波长为550nm,收集最大发射波长600nm处的荧光变化。结果如图5所示,引入2.5%或5%的cpt修饰sp/pgfp纳米复合物,有效提高了纳米复合物的稳定性,且相比sp/dna和bpei/dna,cpt/sp/dna多组分纳米复合物具有更高的稳定性,有助于提高在输送过程中载体材料对dna的保护能力。
性能检测2微量阳离子线性/支化聚噻吩构建的多组份纳米复合物的细胞毒性实验
收集对数生长期hepg2细胞,调整细胞悬液浓度,以每孔大约1.6×104个/200μl的细胞浓度接种到96孔板,置于含有5%co2的37℃培养箱内生长12-24h。当细胞融合70-80%时,将培养基吸除,pbs洗细胞1-2遍,加入含有预设浓度测试样品的完全培养基,继续孵育24h。24h后,吸除含有样品的培养基,pbs洗细胞1-2遍,每孔加入100μl含有mtt(0.5mg/ml)的完全培养基,培养箱内继续孵育4h。4h后吸除培养基,pbs轻洗细胞1-2遍。加入150μldmso,摇床上轻摇20min,使生成的甲瓒晶体充分溶解。酶标仪分别测定各孔在570nm、720nm波长下的吸收值,记录结果。其中,每孔所记录的720nm波长下的吸光度值为扣除背景用,即最终计算用的吸光度值为od570减去od720后的数值。细胞存活率可以按照如下公式计算:
细胞存活率(%)=(样品组平均吸光度值/对照组平均吸光度值)×100%
注意当检测光毒性时,则在多组份纳米复合物体系与细胞共孵育4-6h时引入光照条件即可,其余操作与未光照组一致。
结果如图6所示,sp/pgfp复合物的细胞毒性和cpts构建的多组份纳米复合物体系的细胞毒性基本相近,分子量较低的阳离子聚噻吩材料,在光照及非光照条件下均未产生明显的细胞毒性。
性能检测3微量阳离子线性/支化聚噻吩构建的多组份纳米复合物在细胞内引起ros水平变化测定
细胞内的活性氧可以氧化无荧光的dcfh生成有荧光的dcf,通过检测dcf的荧光强度可以推算出细胞内活性氧水平。将ros捕捉剂dcfh-da用dmso溶解制备成10mm溶液,使用时用pbs稀释至终浓度为20μm。以96孔板为例:收集对数生长期的细胞,调整细胞悬液浓度,以每孔大约1.6×104个/200μl的细胞浓度接种到96孔板,置于含有5%co2的37℃培养箱内生长12-24h。当细胞融合70-80%时,将培养基吸除,pbs洗细胞1-2遍,加入含有载体材料/质粒的纳米复合物的不完全培养基。吸除每孔中含有材料的培养基,pbs洗1-2遍,加入dcfh-da探针溶液(终浓度为20μm),37℃细胞培养箱内避光孵育20min。去除探针溶液,pbs洗2-3遍。洗涤完后每孔加入100μl完全培养基,荧光倒置显微镜下观察记录dcfh-da捕捉了ros后的氧化产物dcf的绿色荧光(ex=465/95nm,em=515-555nm)。
当检测光照作用时,则在多组份纳米复合物体系与细胞共孵育4-6h时引入光照条件即可,其余操作与未光照组一致。
通过图7a、7b至图16a、16b所示,阳离子线性/支化聚噻吩修饰形成的多组份纳米复合物被细胞内化后,在经过白光(500mw/cm2)照射后,源自于dcf的绿色荧光显著增强,说明光照后纳米复合物中的微量cpts能够显著提升细胞内的ros水平,这对增强纳米复合物的溶酶体逃逸具有重要的应用价值。
性能检测4微量阳离子线性/支化聚噻吩构建的多组份纳米复合物被细胞摄取后引起的溶酶体通透性变化。
利用质粒标记试剂盒(miruslabelit),对表达luciferase的质粒pluci进行rhodamine标记,得到rho-pluci。配制含微量cpt和rho-pluci的多组分纳米复合物。实验中,收集对数生长期的细胞,以7×104个/800μl的浓度接种于30mm玻璃培养皿中,置于含有5%co2的37℃培养箱内生长12-24h。当细胞融合70-80%时,将培养基吸除,pbs洗细胞1-2遍,加入含有载体材料/质粒的纳米复合物(质粒终浓度为2μg/ml,n/p5)的不完全培养基。继续孵育4h后,培养皿置于白光(500mw/cm2)下照射1min,对照组细胞则不经受光照。随后使用lysotracker对细胞进行溶酶体染色。激光共聚焦显微镜观察溶酶体荧光探针染料所用的激发波长为488nm,收集了505-525nm波长范围的发射光;观察罗丹明染料所用的激发波长为543nm,收集了560-700nm波长范围的发射光。
结果如图17a、17b、17c、17d、17e、17f至19a、19b、19c、19d、19e、19f所示,对照组pei/dna和sp/dna,在光照或非光照条件下,在细胞内均呈现很强的lysotracker绿色荧光,rho-pluci的红色荧光与lysotracker绿色荧光高度重合,表明溶酶体膜比较完整,纳米颗粒很难从溶酶体逃逸到胞浆。而摄取了微量cpt/sp/dna多组分纳米复合物的细胞,在非光照时,lysotracker绿色荧光明显变暗,表明溶酶体膜完整性受到扰动而导致ph值下降受阻;在光照后,lysotracker的绿色荧光非常暗淡,而且与rho-pluci的红色荧光重合不佳,表明溶酶体膜完整性被破坏、质粒成功从溶酶体逃逸。该结果充分说明利用微量共轭聚合物的pci效应,能够有效辅助纳米复合物的溶酶体逃逸。
性能检测5微量阳离子线性/支化聚噻吩构建的多组份纳米复合物的invitro基因输送实验
收集对数生长期的细胞,调整细胞悬液浓度,以每孔大约1.6×104个/200μl的细胞浓度接种到96孔板,置于含有5%co2的37℃培养箱内生长12-24h。当细胞融合70-80%时,将培养基吸除,pbs洗细胞1-2遍,加入含有载体材料/质粒的纳米复合物的不完全培养基,继续孵育4-6h。随后,吸除含有样品的不完全培养基,pbs洗细胞1-2遍,每孔加入200μl完全培养基,培养箱内继续孵育36-40h。对质粒pgfp的转染效率通过荧光倒置显微镜观察记录(ex=465/95nm,em=515-555nm),对质粒pluc的转染效率通过luciferaseassay(购买于美国promega公司)测定。
当检测光照作用时,则在多组份纳米复合物体系与细胞共孵育4~6h时引入光照条件即可,其余操作与未光照组一致。
阳离子线性/支化聚噻吩构建的多组份纳米复合物体系对hela细胞的pgfp转染40h。结果如图20所示,当转染过程中引入光照条件后,含有微量cpts的基因输送体系对hela细胞的转染效率显著增强。结果表明,相比对照载体材料sp和pei,本发明使用含量为2.5%的cpt、结合pci策略可使luciferase的表达最多增强约10倍以上。
实施例3阳离子聚苯撑乙炔及其制备
本发明的阳离子聚苯撑乙炔的结构式为:
其中,cppe-det合成路线如下所示:
具体的制备方法如下:
(1)2,5-二碘-1,4-二苯乙酸乙酯的合成:将2,5-二碘-1,4-二苯乙酸(4.45g,10mmol)溶于无水乙醇(100ml),滴加二氯亚砜(1ml),室温下搅拌24h,旋蒸除去溶剂,硅胶柱分离后得到浅黄色固体(4.0g);
(2)ppe-et合成:将2,5-二碘-1,4-二苯乙酸乙酯(0.5g,1mmol)溶于无水四氢呋喃(15ml)和三乙胺(5ml),脱气后加入催化剂量的pd(pph3)4和cui,鼓氮气10min,加入三甲基硅烷乙炔(0.27ml,2mmol),室温下反应0.5h,向反应液滴加1m四丁基氟化铵溶液(2ml,2mmol),在室温下继续反应24h,将反应混合物通过短的硅胶柱,用乙醚沉淀3次,得到浅黄色固体(0.16g);
(3)cppe-det的合成:取78mg(0.2mmol)ppe-et溶于10ml干燥的nmp,冷却后,加入二乙基三胺的nmp溶液(100eq,10ml),35℃反应3d,盐酸中和后,先后用盐酸(ph2)和水透析,最后冷冻干燥,得到黄色固体cppe-det。
在制备阳离子聚苯撑乙炔时,步骤(1)中,将2,5-二碘-1,4-二苯乙酸与二氯亚砜的摩尔比替换成10:1、50:1、100:1,均可制得该结构的阳离子聚苯撑乙炔;步骤(2)中,将2,5-二碘-1,4-二苯乙酸乙酯、pd(pph3)4、cui及三甲基硅烷乙炔的摩尔比替换为0.25:0.001:0.001:1、0.5:0.01:0.01:1,均可制得该结构的阳离子聚苯撑乙炔。
实施例4阳离子聚苯撑乙炔构建的多组份纳米复合物
原料组成:该复合物包括聚氨基酸衍生物、rna及阳离子聚苯撑乙炔;其中,阳离子聚苯撑乙炔及聚氨基酸衍生物的正电荷总和与rna的负电荷之比为10:1,共轭聚电解质与聚氨基酸衍生物的正电荷之比为0.1%、0.25%、0.5%或1.0%。
其中,所述聚氨基酸衍生物,为聚赖氨酸或多聚精氨酸,分子量为5~50kda。
制备方法:在聚合物材料所含的胺基(包括1~4级胺)数目与核酸分子所含磷酸酯键数目之比为5的条件下(即n/p=5),首先将聚氨基酸和rna用超纯水稀释到相同的体积后,将rna溶液缓慢加入到聚氨基酸中,完全加完后静置30min,使材料与质粒自组装形成稳定的纳米复合物;随后,向纳米复合物体系中缓慢加入等体积的阳离子聚苯撑乙炔,完全加完后再静置60min,得到稳定的多组份纳米复合物。
性能检测6微量阳离子聚苯撑乙炔构建的多组份纳米复合物的invitro基因输送实验
收集对数生长期的细胞,调整细胞悬液浓度,以每孔大约1.6×104个/200μl的细胞浓度接种到96孔板,置于含有5%co2的37℃培养箱内生长12~24h;当细胞融合70-80%时,将培养基吸除,pbs洗细胞1~2遍,加入含有载体材料/质粒的纳米复合物的不完全培养基,继续孵育4~6h;随后,吸除含有样品的不完全培养基,pbs洗细胞1-2遍,每孔加入200μl完全培养基,培养箱内继续孵育36-40h,对pgfp的转染水平通过荧光倒置显微镜观察记录(ex=465/95nm,em=515-555nm),对pluci的转染效率通过luciferaseassaykit(promega公司)定量测定。
注意当检测光照作用时,则在多组份纳米复合物体系与细胞共孵育4-6h时引入光照条件即可,其余操作与未光照组一致。
微量阳离子聚苯撑乙炔的多组份纳米复合物体系对hela细胞转染40h的结果如图21a、21b至26a、26b及图27所示,cppe修饰后能够显著提高sp/dna复合物对pgfp和pluc转染效率。0.1%、0.25%、0.5%、1.0%不同的cppe含量条件下转染效率均有提升,0.25%的作用效果最佳。luciferaseassay检测结果显示,在不引入光照的条件下,pim-2就具有很强的促进sp/dna复合物转染的能力,0.25%的pim-2材料加入后,多组份纳米复合物的基因转染效率相比sp提高了10倍,相比bpei提高了约23倍。上述结果均证实了微量阳离子聚苯撑乙炔构建的多组份纳米复合物体系能够显著提高基因输送效率。
实施例5阳离子聚苯撑乙烯及其制备
本发明的阳离子聚苯撑乙烯的结构式为:
其中,n=10~200。
其制备路线为:
制备方法包括如下步骤:
(1)2,5-二碘-1,4-二苯乙酸乙酯的合成:将2,5-二碘-1,4-二苯乙酸(4.45g,10mmol)溶于无水乙醇(100ml),滴加二氯亚砜(1ml),室温下搅拌24h,旋蒸除去溶剂,硅胶柱分离后得到浅黄色固体(4.0g);
(2)ppv-et的合成:将2,5-二碘-1,4-二苯乙酸乙酯(0.5g,1mmol)、1,4-苯二乙烯(0.13g)溶于无水二氧六环(10ml),鼓氮气30min;氮气气氛下加入三乙胺(1ml)和催化量5%pd/c,加热回流,反应24h,过滤后旋蒸除去溶剂,氯仿溶解,在乙醚中沉淀3次,得到黄色固体(0.3g);
(3)cppv-det的合成:取78mg(0.2mmol)ppv-et溶于10ml干燥的nmp,冷却后,加入二乙基三胺的nmp溶液(100eq,10ml),35℃反应3d;盐酸中和后,先后用盐酸(ph2)和水透析,最后冷冻干燥,得到黄色固体cppv-det。
在制备阳离子聚苯撑乙烯时,步骤(1)中,将2,5-二碘-1,4-二苯乙酸与二氯亚砜的摩尔比1替换成10:1、50:1、100:1,均可制得该结构的阳离子聚苯撑乙烯;步骤(2)中,将2,5-二碘-1,4-二苯乙酸乙酯与1,4-苯二乙烯的摩尔比替换为0.8:1、1:1、1.25:1,均可制得该结构的阳离子聚苯撑乙烯。
实施例6阳离子聚苯撑乙烯构建的多组份纳米复合物
原料组成:该复合物包括聚酰胺衍生物、dna及阳离子聚苯撑乙烯;其中,阳离子聚苯撑乙烯及聚酰胺衍生物的正电荷总和与dna的负电荷之比为1:1,阳离子聚苯撑乙烯与聚酰胺衍生物的正电荷之比为0.01%。其中,所述聚酰胺衍生物为胺型聚酰胺pamam(第3.0~6.0代)。
制备方法:在聚合物材料所含的胺基(包括1~4级胺)数目与核酸分子所含磷酸酯键数目之比为5的条件下(即n/p=5),首先将聚酰胺衍生物和dna用超纯水稀释到相同的体积后,将质粒溶液缓慢加入到聚酰胺衍生物中,完全加完后静置30min,使材料与质粒自组装形成稳定的纳米复合物。随后,向纳米复合物体系中缓慢加入等体积的阳离子聚苯撑乙烯,完全加完后再静置30min,得到稳定的多组份纳米复合物。
实施例7阳离子共聚物聚(芴-co-苯)及其制备
本发明的阳离子共聚物聚(芴-co-苯)的结构式为:
其中,n=10~200。
其制备路线为:
具体的制备方法包括如下步骤:
将fl-et(496mg,1mmol)、1,4-二苯硼酸(166mg,1mmol)溶于甲苯(10ml)和碳酸钾水溶液(5ml,2m),脱气后加入催化量的四(三苯基膦)钯,90℃反应24h,氯仿萃取、水洗、无水硫酸钠干燥,旋蒸浓缩,再经3次乙醚沉淀、干燥,得到浅黄色固体pfp-et(220mg),取82mg(0.2mmol)pfp-et溶于10ml干燥的nmp,冷却后,加入r-nh2的nmp溶液(100eq,10ml),35℃反应3d,盐酸中和后,先后用盐酸(ph2)和水透析,最后冷冻干燥,得到黄色固体cpfp-r。
在制备阳离子共聚物聚(芴-co-苯)时,将fl-et、1,4-二苯硼酸与四(三苯基膦)钯的摩尔比为替换为10:10:1,25:25:1,均可制得该结构的阳离子共聚物聚(芴-co-苯)。
实施例8阳离子共聚物聚(芴-co-苯)构建的多组份纳米复合物
原料组成:该复合物包括聚糖衍生物、rna及阳离子聚(芴-co-苯);其中,阳离子聚芴及聚糖衍生物的正电荷总和与rna的负电荷之比为20:1,阳离子聚(芴-co-苯)与聚糖衍生物的正电荷之比为10%。其中,所述聚糖衍生物为壳聚糖。
制备方法:在聚合物材料所含的胺基数目与核酸分子所含磷酸酯键数目之比为5的条件下(即n/p=5),首先将聚糖和rna用超纯水稀释到相同的体积后,将质粒溶液缓慢加入到聚糖中,完全加完后静置20min,使材料与质粒自组装形成稳定的纳米复合物,随后,向纳米复合物体系中缓慢加入等体积的阳离子聚(芴-co-苯),再静置10min,得到稳定的多组份纳米复合物。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种TaC@洋葱状碳/无定形碳纳米复合物及其制备方法和应用与制造工艺
- 一种W2C@洋葱状碳/无定形碳纳米复合物及其制备方法和应用与制造工艺
- 五氧化二矾及其碳纳米复合物规模化制备与锂电池应用的制造方法与工艺
- 一种ZnPc‑UCNP‑PEG‑G纳米复合物的制备方法与制造工艺
- 蒙脱土增强固体丙烯酸酯树脂及其制备方法与制造工艺
- 一种Fe(Ⅲ)和Cu(Ⅱ)掺杂的聚苯胺纳米复合物及其应用
- 一种Sn@C@g?C<sub>3</sub>N<sub>4</sub>纳米复合物及其制备方法
- 一种Co@C@g?C<sub>3</sub>N<sub>4</sub>纳米复合物及其制备方法和应用
- 基于超支化纳米复合物的双酚a增敏型免疫层析金标试纸条的制备方法
- 一种的强韧性石墨烯纳米改性橡胶及其制备工艺的制作方法
- 一种基于生物功能化纳米银装载紫杉醇或其类似物的靶向递送系统的制作方法与工艺
- 一种Al‑Zn‑Si纳米复合物速凝早强剂及其制备方法与流程
- 一种载纳米银的PNIPAM/PVA复合温敏凝胶的制备及其应用的制作方法与工艺
- 一种利用蓝莓叶提取液生物合成的纳米银抑菌剂及其制备工艺的制作方法与工艺
- 金属‑有机配合物与银纳米线复合物及其制备方法与应用与流程
- 一种TiO2/SnO2/Carbon纳米复合物及其制备方法与流程
- 一种含纳米NaY分子筛的复合物及其制备方法与流程
- 基于CdTe‑Ag2Se纳米复合物的皮质酮电化学发光传感器的制备方法和应用与流程
- 一种以纳米银茶叶渣生物碳为主滤料的车载式空气净化器的制作方法与工艺
- PLGA纳米复合物及其制备方法与流程