一种简便高效合成地诺前列素的方法与流程
本发明涉及医药制备领域,特别涉及一种简便高效合成地诺前列素的方法。
背景技术:
前列腺素(prostaglandins,简称pgs)是一类具有广泛生理活性的重要内源性产物。pgs最早由美国学者voneluer于1930年发现并命名,1962年,bergstorm提取出两种pg纯品(pgfl和pgf2。),并确定其化学结构。1969年willis首次提出pgs是体内的一种炎症介质。随后pgs的各种生理活性、药理活性受到深入研究。它存在于几乎所有哺乳动物的组织中,在生殖系统、消化系统、呼吸系统和心血管系统中发挥着重要作用,参与体温调节、炎症反应、青光眼、妊娠、高血压、溃疡及哮喘等生理病理过程。pgs的结构特征如下:具有一个五元脂环及两个侧链,上侧链7个碳、下侧链8个碳。目前工业生产主要采用corey内酯及其衍生物为原料的合成方法,其特点在于节约成本,也具有一定工业应用前景。
地诺前列素(inn),简称为pgf2α,它在医学上的用途是作为天然的前列腺素用于引产和堕胎药。通常是由哺乳动物子宫受刺激而产生的,其作用于黄体导致黄体退化,形成了纤维化的卵巢白体。pgf2α的运动是根据黄体模受体0的数目而进行。该pgf2α亚型8-异前列腺素f2α被发现在子宫内膜异位症患者有显著的增加,因而是一种与子宫内膜异位相关的氧化应激潜在因子。
在20世纪70年代,以corey为代表的化学家们完成了pgs的人工化学全合成工作。随后世界各国研究者们也纷纷投入对前列素的合成研究中,但进展缓慢。在最近十几年里,对前列腺素类化合物的化学合成研究有了较大的突破。有关合成pgs及其类似物的报道大量涌现,其中尤以金属催化的不对称合成方法最为突出。
目前化学合成地诺前列素的方法主要是以左旋科里内酯为原料,经过九步反应制备,总收率约在15%-20%,如图1所示。该工艺经过选择性保护伯醇、仲醇及脱保护才得到重要中间体——苯甲酰科里内酯,然后再经过系列化学反应,得到终产物。因此导致其缺点较多:步骤长,收率低,成本高;同时,中间体纯化方法和杂质控制也是其难以实现工业化的壁垒。
中国专利申请-“氨基丁三醇前列腺素f2α的合成方法”(公开日:2017年6月9日)中涉及前列腺素f2α的合成制备方法,该制备方法制备前列腺素f2α时,收率低,操作复杂。
技术实现要素:
为解决上述问题,本发明提供一种简便高效合成地诺前列素的方法,具体合成步骤如下:
s110:以dmn-azado为催化剂,将左旋科里内酯二醇选择性氧化为含醛基的科里内酯单醇——化合物i;
s120:以氢氧化锂为催化剂,化合物i与2-氧代庚基膦酸二甲酯发生霍纳尔-沃兹沃思-埃蒙斯反应,生成反式产物化合物ii;
s130:以硼氢化锌为还原剂,在dme溶剂中,以s-binap为手性诱导催化剂,手性还原共轭酮羰基至手性醇,随后以路易斯酸三氟化硼乙醚溶液为催化剂将内酯还原为醇,得中间体——化合物iii;
s140:将s130所得中间体iii选择性氧化伯醇,得中间体——化合物iv;
s150:将化合物iv与上侧链中间体brpph3(ch2)4cooh,加入nahmds,以hmpa为反应溶剂离子捕获剂,经过witting反应,生成地诺前列素;
其中:氧化左旋科里内酯二醇的结构式为
进一步地,s110中的催化剂dmn-azado的用量为1~10mol%;反应温度为0~30℃。
进一步地,s110中使用的共氧化剂碘苯乙酸酯、次氯酸钠、氯酸钠、溴化钾、四丁基溴化铵中的一种或者多种。
进一步地,s110中使用的反应溶剂是二氯甲烷或者乙腈的一种或者多种;反应溶剂使用量为反应底物的0.1~1mol/l。
进一步地,s120中以氢氧化锂为催化剂,以乙腈、四氢呋喃或者二氯甲烷的一种或者多种为反应溶剂;其中氢氧化锂使用当量为反应底物的1.5~3倍;反应溶剂为0.1~1mol/l;反应温度为10~30℃。
进一步地,s130中以dme为溶剂,在s-(-)-binap协同催化下,使用zn(bh4)2还原剂,得到化合物iii的中间体,再经过dibal-h还原内酯制得化合物iii;其中,s-(-)-binap使用当量为1~10mol%;反应温度为-20~-10℃。
进一步地,s130中手性醇还原结束后,以三氟化硼乙醚溶液为催化剂,采用一锅法还原内酯,生成伯醇化合物iii。
进一步地,s140中以dmn-azado为催化剂选择性氧化伯醇生成化合物iv;其中:dmn-azado的用量为1~10mol%;反应温度为0~30℃。
进一步地,s150中nahmds使用当量为底物的3.5~12倍;hmpa使用浓度为0.1~1mol/l;反应温度为-40~-20℃。
cn201611236550公开了一种氨基丁三醇前列腺素f2α的合成方法。本发明提供的简便高效合成地诺前列素的方法,与其相比具有明显的优势:
1、cn201611236550公开的氨基丁三醇前列腺素f2α的合成方法使用四甲基哌啶氮氧化物及二乙酸碘苯为氧化剂,导致伯醇和仲醇氧化时反应选择性不高;
2、cn201611236550公开的氨基丁三醇前列腺素f2α的合成方法拼接下侧链步骤繁琐,需要分三次加入下侧链、丙酮、水及碳酸钾,较难实现工业化,且收率只有43.8%,而采用本发明提供的简便高效合成地诺前列素的方法,操作简单,两步收率90%;
3、本发明提供的简便高效合成地诺前列素的方法使用相对温和的试剂,代替dibal-h作为反应还原剂,使得反应无需在低温下进行,却可以得到更好的反应效果,同时反应中间体是固体,重结晶即可实现产品的纯化,最关键是dibal-h还原无法控制反应选择性,不能得到单一的手性醇化合物;
4、本发明提供的简便高效合成地诺前列素的方法拼接上侧链时,反应收率明显提高;cn201611236550公开的氨基丁三醇前列腺素f2α的合成方法拼接上侧链时,单步反应收率只有54%,而本文整个工艺收率为50-60%;
5、cn201611236550公开的氨基丁三醇前列腺素f2α的合成方法使用大量的乙腈为成盐溶剂,本发明提供的简便高效合成地诺前列素的方法则以异丙醇代替乙腈,可以大大的减少溶剂成本和操作流程。
本发明提供的一种简便高效合成地诺前列素的新方法,以左旋科里内酯为起始原料,通过选择性氧化、拼接下侧链、选择性连续还原、拼接上侧链等步骤,制备高纯度地诺前列素,反应收率可达到55-60%。该方法优点在于:原料简单易得,设备要求简单;步骤简短,操作方便且无需-78℃底温反应,容易实现工业化生产;整个过程中没有使用到重金属或者其他强致癌物,可以有效的扩大产品的使用范围;中间体纯化方法简单、杂质容易控制,得到的产物光学纯度高等优点。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作一简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动性的前提下,还可以根据这些附图获得其他的附图。
图1为本发明提供的简便高效合成地诺前列素的新方法路线图;
图2为pgf2α的c谱图;
图3为pgf2α的h谱图;
图4为化合物i的c谱图;
图5为化合物i的h谱图;
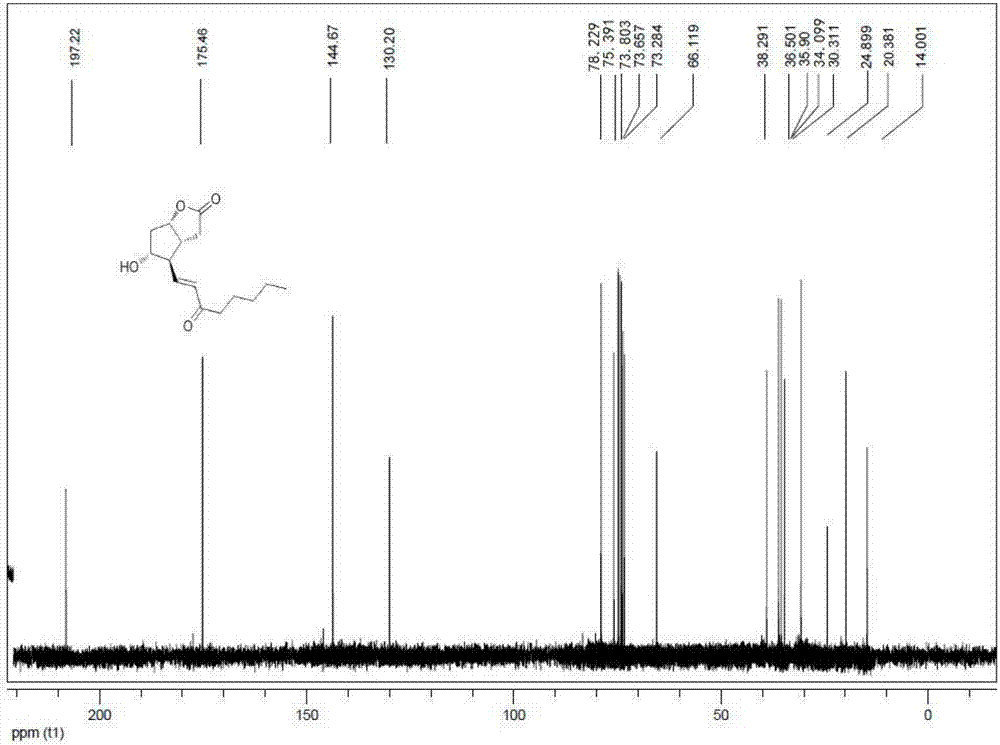
图6为化合物ii的c谱图;
图7为化合物ii的h谱图。
具体实施方式
为使本发明实施例的目的、技术方案和优点更加清楚,下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
本发明实施例提供一种简便高效合成地诺前列素的方法,具体合成步骤如下:
s110:以dmn-azado为催化剂,将左旋科里内酯二醇选择性氧化为含醛基的科里内酯单醇——化合物i;dmn-azado由于具有很大的空间位阻,只能配位氧化伯醇,因此选择性和收率都高;
s120:以氢氧化锂为催化剂,化合物i与2-氧代庚基膦酸二甲酯发生霍纳尔-沃兹沃思-埃蒙斯反应,生成反式产物化合物ii;氢氧化锂作为碱,与2-氧代庚基膦酸二甲酯反应生成负离子,又作为反应中间体的络合金属,有效的促进反应的进行;
s130:以硼氢化锌为还原剂,在dme溶剂中,以s-binap为手性诱导催化剂,手性还原共轭酮羰基至手性醇,随后以路易斯酸三氟化硼乙醚溶液为催化剂将内酯还原为醇,得中间体——化合物iii;
将s-binap作为手性配体,先与硼氢化锌形成中间体手性络合,再与反应底物反应;由于空间位阻效应,只能选择性的还原;
s140:将s130所得中间体iii选择性氧化伯醇,得中间体——化合物iv;
s150:将化合物iv与上侧链中间体brpph3(ch2)4cooh,加入nahmds,以hmpa为反应溶剂离子捕获剂,可以稳定的捕获上述中间体,促进反应单向进行,提高反应速度和收率,经过witting反应,生成地诺前列素;
其中,nahmds是一种碱,与brpph3(ch2)4cooh反应生成中间体pph3ch-(ch2)3coo-;
其中:氧化左旋科里内酯二醇的结构式为
优选地,s110中的催化剂dmn-azado的用量为1~10mol%;反应温度为0~30℃。
s110反应中,与传统的方法相比较,科里内酯二醇不需要经过系列的保护和脱保护再氧化伯醇,而是利用新型催化剂dmn-azado直接选择性氧化伯醇生成化合物i;其催化剂使用量1-10mol%;反应温度为0~30℃,但不局限于该温度或者某温度点;该方法选择性专一,收率高,且得到的粗产品无需进一步纯化,即可以直接应用于下一步反应。
优选地,s110中使用的共氧化剂碘苯乙酸酯、次氯酸钠、氯酸钠、溴化钾、四丁基溴化铵中的一种或者多种。
优选地,s110中使用的反应溶剂是二氯甲烷或者乙腈的一种或者两者的混合物;反应溶剂使用量为反应底物的0.1~1mol/l。
优选地,s120中以氢氧化锂为催化剂,以乙腈、四氢呋喃或者二氯甲烷的一种或者或者混合;其中氢氧化锂使用当量为反应底物的1.5~3倍;反应溶剂为0.1~1mol/l;反应温度为10~30℃。
s120反应中,以氢氧化锂为催化剂,以乙腈、四氢呋喃或者二氯甲烷为溶剂,单独或者混合使用;其中氢氧化锂使用当量为反应底物的1.5~3倍;反应溶剂为0.1~1mol/l;反应温度为10~30℃;与以氯化锂为反应催化剂相比较,氢氧化锂具有选择性高,反应条件温和,无使用有机碱,后处理简单等优点。
优选地,s130中以dme为溶剂,在s-(-)-binap协同催化下,使用zn(bh4)2还原剂,得到化合物iii的中间体;其中,s-(-)-binap使用当量为1~10mol%;反应温度为-20~-10℃。
优选地,s130中手性醇还原结束后,以三氟化硼乙醚溶液为催化剂,采用一锅法还原内酯,生成伯醇化合物iii。
优选地,s140中以dmn-azado为催化剂选择性氧化伯醇生成化合物iv;其中:dmn-azado的用量为1~10mol%;反应温度为0~30℃。
s140选择性氧化采用和s110相同反应条件,利用新型催化剂dmn-azado直接选择性氧化伯醇生成化合物iv;其催化剂使用量1~10mol%;反应温度为0~30℃,但不局限于该温度或者某温度点;得到重要的拼接中间体,化合物iv。
优选地,s150中nahmds使用当量为底物的3.5~12倍;hmpa使用浓度为0.1~1mol/l;反应温度为-40~-20℃。
s140得到的化合物iv,无需要经过纯化,即可以直接进行s150中的反应,而且收率可以达到90%;nahmds使用当量为底物的3.5~12倍,hmpa作为反应溶剂和离子捕获剂,可以有效的提高反应收率,其使用浓度为0.1~1mol/l;反应温度为-40~-20℃。
实施例
具体合成步骤如图1所示:
步骤(一)化合物i制备
在装有恒压滴液漏斗、温度计及机械搅拌的10l三口瓶中分别加入科里内酯二醇(500g,2.907mol),dmn-azado(4.93g,0.02907mol)、溴化钾kbr(34.59g,0.2907mol)、n-bu4nbr(47.8g,0.1454mol)、二氯甲烷(5l)及饱和碳酸氢钠溶液(400ml);然后将上述混合体系使用冰盐浴,冷却至内温≤0℃,并恒温搅拌10min;然后将naclo(3.4884mol,用作氧化剂)溶液缓慢滴加至反应体系,控制反应温度不高于5℃;滴加结束,再恒温搅拌30min。反应结束后,体系中加入饱和na2s2o3(中和过量的氧化剂)(1000ml)猝灭反应,水相使用二氯甲烷萃取(0.5l*2)。合并有机相再使用饱和氯化钠溶液洗涤、干燥、浓缩,得淡黄色化合物i(501g,纯度97%),可直接应用于下步反应。产物的谱图如图4和图5所示。1hnmr(400mhz,cdcl3):δ9.71(s,1h),5.03(t,j=6.7hz,1h),4.55-4.59(m,1h),3.32-3.40(m,1h),2.89-2.96(m,2h),2.51(dd,j=9.2,3.5hz,1h),2.10-2.17(m,1h),1.81-1.89(m,1h);13cnmr(100mhz,cdcl3):δ199.5,176.7,84.3,73.8,68.1,41.1,36.5,35.9。
步骤(二)化合物ii制备
在装有温度计、机械搅拌及滴液漏斗的10l的反应釜中,顺次加入lioh·h2o(293g,6.977mol)和乙腈(2l)。开启搅拌,室温搅拌5min。接着将下侧链2-氧代庚基膦酸二甲酯(968g,4.36mol)溶解于3l的乙腈中,并缓慢滴加至上述体系,30min滴完,再室温搅拌1h;然后再将步骤一中得到的中间化合物i溶解于1l的二氯甲烷中,缓慢的滴加至上述反应体系中,滴加结束,室温搅拌1h;hplc监控反应结束后,加入饱和氯化钠溶液3l,水相再使用乙酸乙酯萃取(2l*2);合并有机相,分别经过饱和氯化钠洗涤、无水硫酸钠干燥、浓缩后得淡黄色,化合物ii(701g,纯度98%),两步收率90%。产物的谱图如图6和图7所示。1hnmr(400mhz,cdcl3):δ(ppm):6.53(1h,dd,j=16,7.5hz),6.14(1h,dd,j=16hz),5.01-5.08(1h,m),4.99-4.86(1h,m),2.81-2.25(5h,m),2.20(2h,j=7.0hz),2.18-2.05(1h,m),1.50-1.34(2h,m),1.20-1.00(4h,m),0.70(3h,t,j=7.5hz);13cnmr(100mhz,cdcl3):δ197.2,175.5,144.7,130.2,78.2,75.4,66.1,38.3,36.5,35.9,34.1,30.3,24.9,20.4,14.0。
步骤(三)化合物iii制备
在10l的反应釜中,加入s-(-)-binap(81.3g,0.13mol)、化合物ii(701g,2.616mol)及无水dme(4l)。将反应釜内温度降至-15℃后,分批次缓慢的加入硼氢化锌(425g,6.54mol),控制加入速度,维持反应温度-15至-10℃。加完后,再恒温反应24h。hplc检测反应结束后。然后再缓慢的滴加46.5%的三氟化硼乙醚溶液(439g,3mol),控制滴加速度,使反应温度不高于0℃。滴加结束,再室温反应1h。缓慢的将100ml甲醇加入至上述反应体系,猝灭反应。再加入5l饱和氯化铵溶液,并使用乙酯乙酯萃取(1l*3),合并有机相,分别使用饱和氯化钠洗涤、无水硫酸钠干燥、浓缩后得粗品654g,收率92%。
步骤(四)化合物iv制备
在装有恒压滴液漏斗、温度计及机械搅拌的10l三口瓶中分别加入化合物iii(654g,2.387mol),dmn-azado(4.04g,0.02387mol)、溴化钾kbr(28.40g,0.2387mol)、n-bu4nbr(39.20g,0.1192mol)、二氯甲烷(5l)及饱和碳酸氢钠溶液(400ml);然后将上述混合体系使用冰盐浴,冷却至内温≤0℃,并恒温搅拌10min;然后将naclo(2.86mol)溶液缓慢滴加至反应体系,控制反应温度不高于5℃;滴加结束,再恒温搅拌30min。反应结束后,体系中加入饱和na2s2o3(1000ml)猝灭,水相使用二氯甲烷萃取(0.5l*2)。合并有机相再使用饱和氯化钠溶液洗涤、干燥、浓缩,得淡黄色化合i(650g,纯度97%),可直接应用于下步反应。
步骤(五)地诺前列素的制备
在装有温度计、机械搅拌及滴液漏斗的20l的反应釜中,顺次加入上侧链中间体溴化戊酸三苯基膦鎓,即:brpph3(ch2)4cooh(4309g,9.705mol)、hmpa(500ml)和四氢呋喃(6l);开启搅拌,将反应釜内温度降至-40℃,再缓慢的将nahmds(9.705mol,4.85l)滴加至反应体系,并恒温反应30min,接着再升温至-25℃,搅拌1h;然后再将步骤四中得到的中间化合物iv(650g,纯度92%),溶解于1.2l的四氢呋喃中,缓慢的滴加至反应体系,滴加结束,再恒温搅拌1h。然后缓慢回温至室温,搅拌过夜;反应结束后,缓慢的加入饱和氯化钠溶液4l,静置分层,水相再使用乙酸乙酯萃取(1l*3),弃去;所得水相再利用15%磷酸酸化至ph=2,再使用乙酸乙酯萃取(1l*3),合并有机相,分别经过饱和氯化钠洗涤、无水硫酸钠干燥、浓缩后得淡黄色液体1214g;再加入3l异丙醇和丙酮(10:3),搅拌均匀后冷冻析晶;晶体再使用丙酮/二氯甲烷溶液淋洗,得粗品化合物(623g,纯度92%)。
将得到的粗品化合物(623g,92%),溶解于异丙醇(1.25l)中,室温搅拌5min;然后将乙酸乙酯/石油醚混合液(2.5l,1:3),缓慢的滴加至上述体系,并置于-10℃下,搅拌析晶12h。将晶体过滤后,滤饼再使用乙酸乙酯/石油醚混合液(1l,1:3)淋洗、干燥,得白色固体pgf2α(544g,纯度≥98.5%),反应总收率52.1%。产物的谱图如图2和图3所示[α]d+28.0(c0.15,chcl3);ir(film)3394,1709cm-1;1hnmr(400mhz,dmso-d6):δ(ppm):5.45(dd,j=15.4,10.6hz,1h),5.36(dd,j=15.6,6.8hz,1h),5.24–5.28(m,2h),5.1-4.0(bs,12h)3.91–3.84(m,3h),2.12–2.05(m,8h),1.53–1.41(m,4h),1.35–1.24(m,8h),0.86(t,j=6.8hz,3h);13cnmr(100mhz,dmso-d6)δ175.8,135.9,132.2,129.9,129.3,76.2,71.7,69.9,62.3,58.7,54.7,49.4,44.4,38.0,34.9,31.8,26.8,25.5,25.2,25.1,22.6,14.4。
将白色固体pgf2α放入氨基丁三醇溶液,冷却过滤得到晶体,晶体用乙腈洗涤后,真空干燥至恒重,得到氨基丁三醇前列腺素f2α。
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
- 还没有人留言评论。精彩留言会获得点赞!